

The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity
- PMID: 25684150
- PMCID: PMC4409520
- DOI: 10.1002/humu.22768
The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity
Abstract
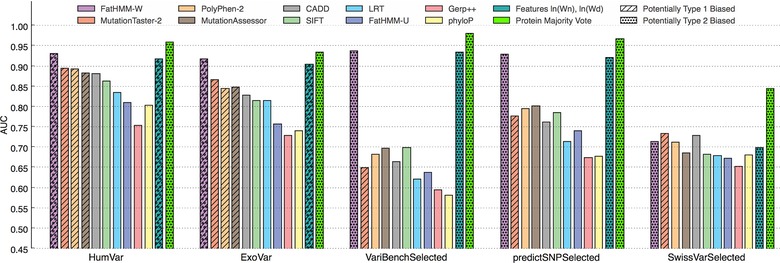
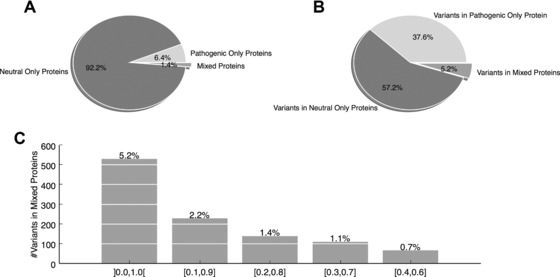
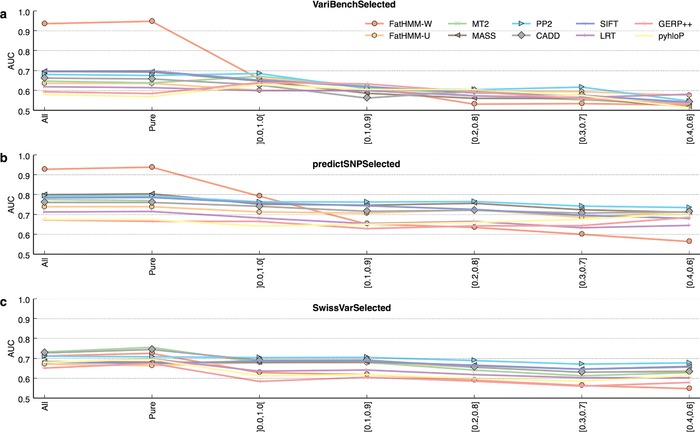
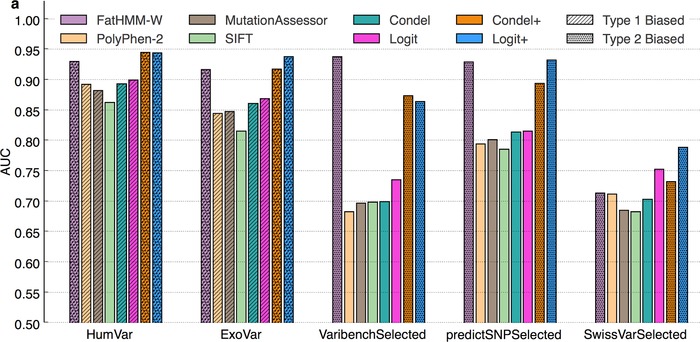
Prioritizing missense variants for further experimental investigation is a key challenge in current sequencing studies for exploring complex and Mendelian diseases. A large number of in silico tools have been employed for the task of pathogenicity prediction, including PolyPhen-2, SIFT, FatHMM, MutationTaster-2, MutationAssessor, Combined Annotation Dependent Depletion, LRT, phyloP, and GERP++, as well as optimized methods of combining tool scores, such as Condel and Logit. Due to the wealth of these methods, an important practical question to answer is which of these tools generalize best, that is, correctly predict the pathogenic character of new variants. We here demonstrate in a study of 10 tools on five datasets that such a comparative evaluation of these tools is hindered by two types of circularity: they arise due to (1) the same variants or (2) different variants from the same protein occurring both in the datasets used for training and for evaluation of these tools, which may lead to overly optimistic results. We show that comparative evaluations of predictors that do not address these types of circularity may erroneously conclude that circularity confounded tools are most accurate among all tools, and may even outperform optimized combinations of tools.
Keywords: exome sequencing; pathogenicity prediction tools.
© 2015 The Authors. **Human Mutation published by Wiley Periodicals, Inc.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
