

Improved data analysis for the MinION nanopore sequencer
- PMID: 25686389
- PMCID: PMC4907500
- DOI: 10.1038/nmeth.3290
Improved data analysis for the MinION nanopore sequencer
Abstract
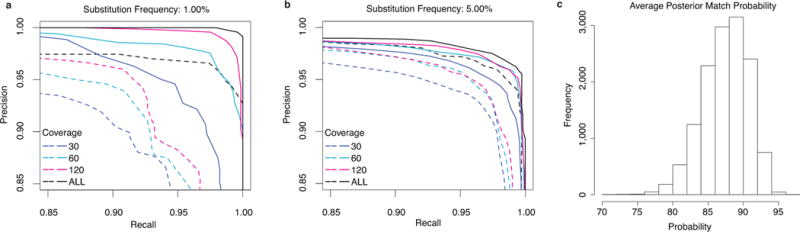
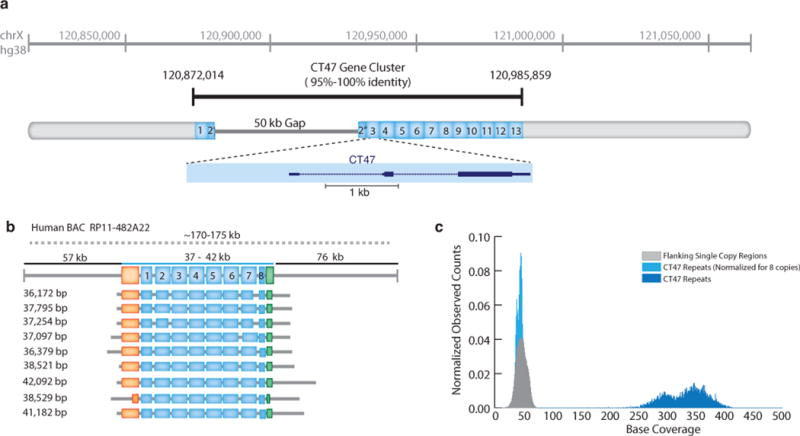
Speed, single-base sensitivity and long read lengths make nanopores a promising technology for high-throughput sequencing. We evaluated and optimized the performance of the MinION nanopore sequencer using M13 genomic DNA and used expectation maximization to obtain robust maximum-likelihood estimates for insertion, deletion and substitution error rates (4.9%, 7.8% and 5.1%, respectively). Over 99% of high-quality 2D MinION reads mapped to the reference at a mean identity of 85%. We present a single-nucleotide-variant detection tool that uses maximum-likelihood parameter estimates and marginalization over many possible read alignments to achieve precision and recall of up to 99%. By pairing our high-confidence alignment strategy with long MinION reads, we resolved the copy number for a cancer-testis gene family (CT47) within an unresolved region of human chromosome Xq24.
Conflict of interest statement
MA is a consultant to Oxford Nanopore Technologies.
Figures
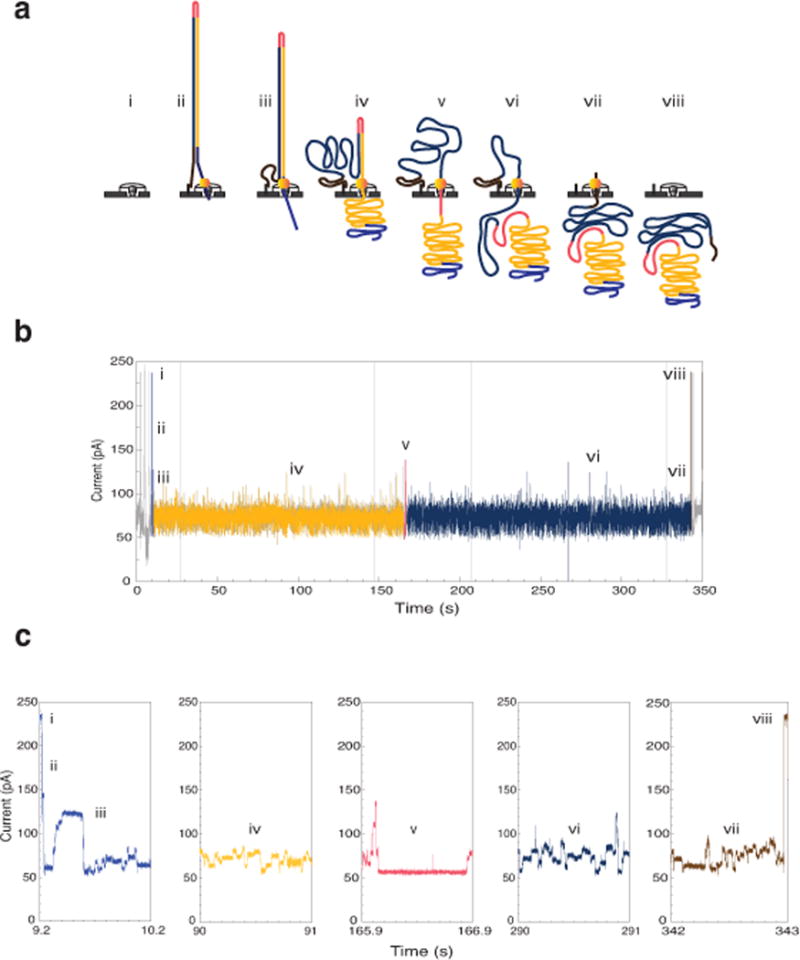
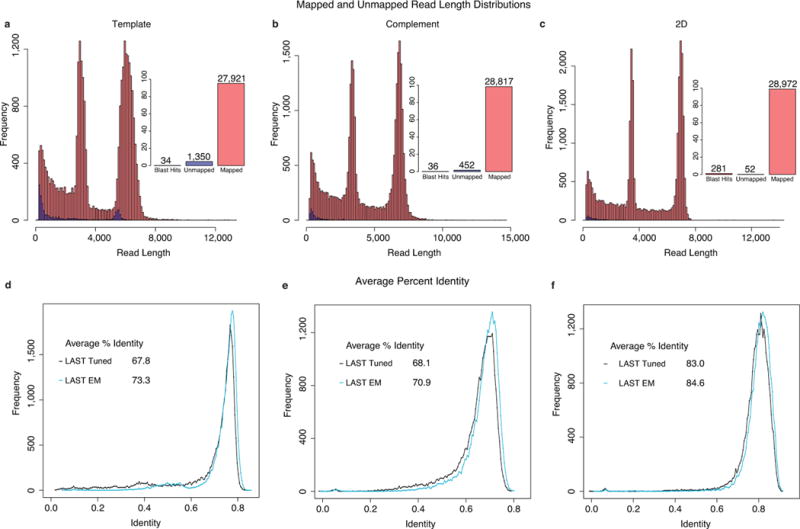
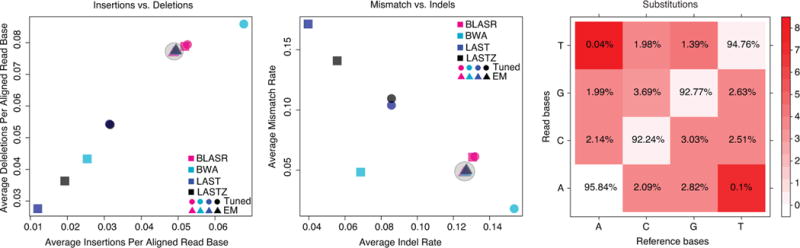
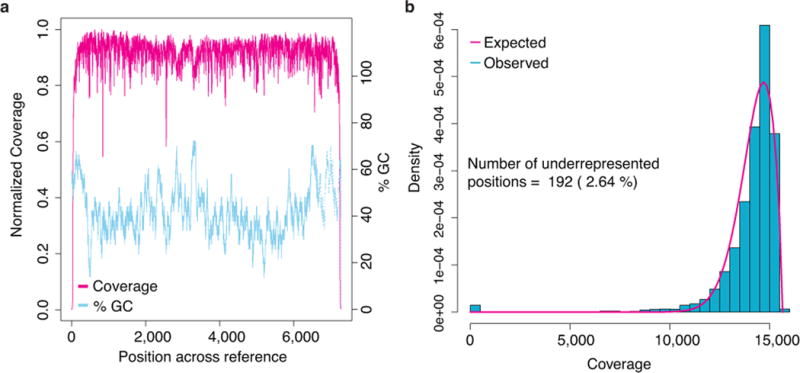
Comment in
-
Successful test launch for nanopore sequencing.Nat Methods. 2015 Apr;12(4):303-4. doi: 10.1038/nmeth.3327. Nat Methods. 2015. PMID: 25825834 No abstract available.
References
-
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013;00:3.
-
- Harris RS. Ph.D. thesis. The Pennsylvania State University; 2007. Improved pairwise alignment of genomic DNA.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
