

The role of inflammation in perinatal brain injury
- PMID: 25686754
- PMCID: PMC4664161
- DOI: 10.1038/nrneurol.2015.13
The role of inflammation in perinatal brain injury
Abstract
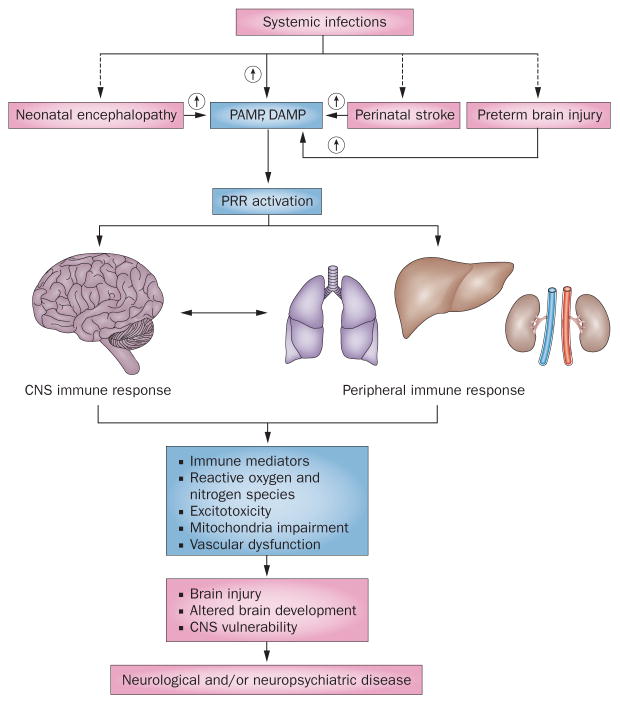
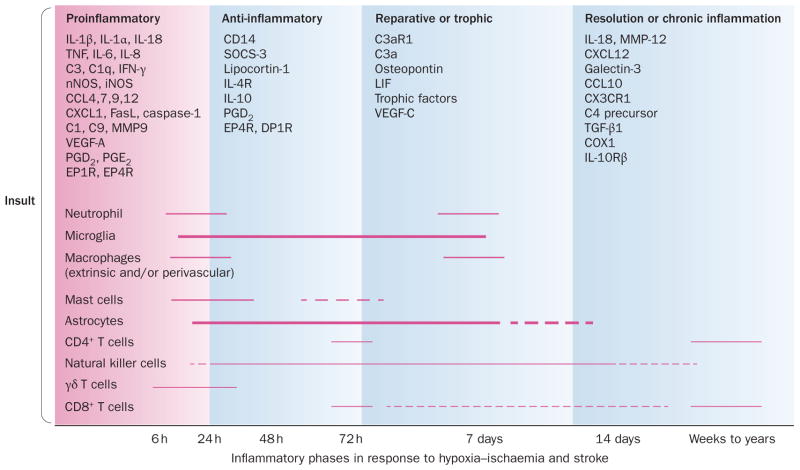
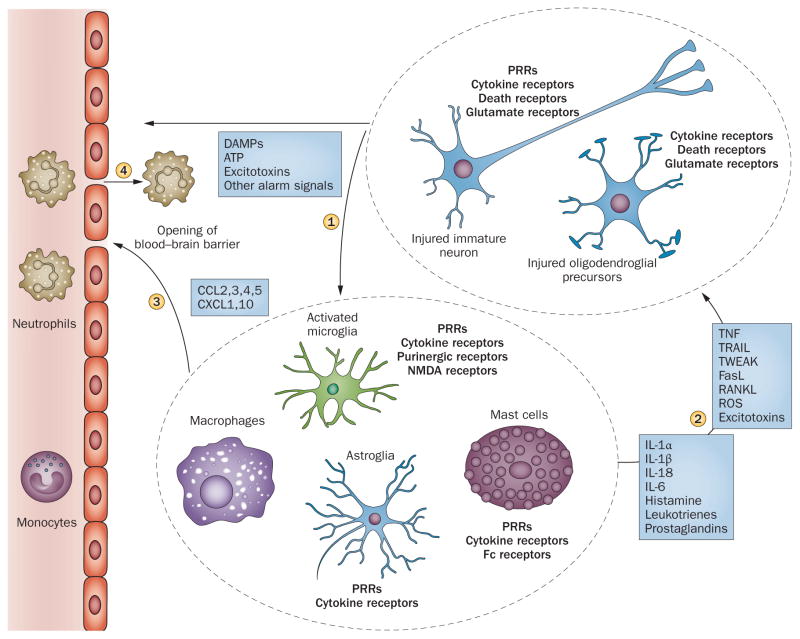
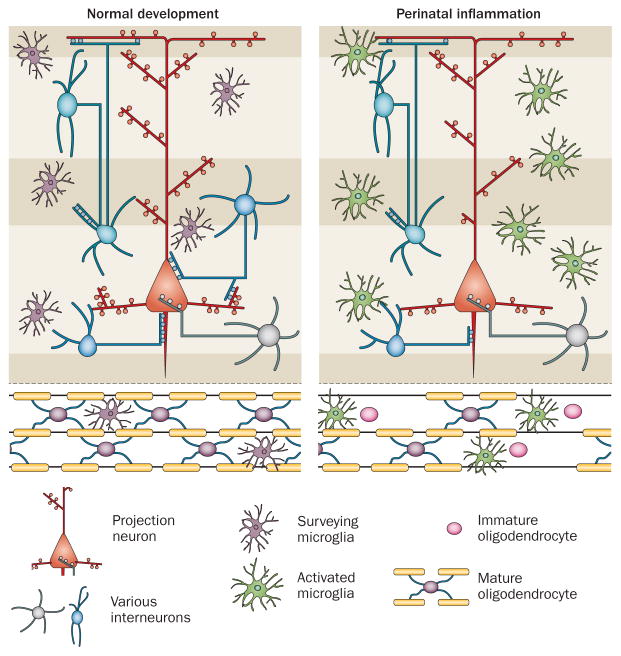
Inflammation is increasingly recognized as being a critical contributor to both normal development and injury outcome in the immature brain. The focus of this Review is to highlight important differences in innate and adaptive immunity in immature versus adult brain, which support the notion that the consequences of inflammation will be entirely different depending on context and stage of CNS development. Perinatal brain injury can result from neonatal encephalopathy and perinatal arterial ischaemic stroke, usually at term, but also in preterm infants. Inflammation occurs before, during and after brain injury at term, and modulates vulnerability to and development of brain injury. Preterm birth, on the other hand, is often a result of exposure to inflammation at a very early developmental phase, which affects the brain not only during fetal life, but also over a protracted period of postnatal life in a neonatal intensive care setting, influencing critical phases of myelination and cortical plasticity. Neuroinflammation during the perinatal period can increase the risk of neurological and neuropsychiatric disease throughout childhood and adulthood, and is, therefore, of concern to the broader group of physicians who care for these individuals.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. - PubMed
-
- Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol. 2012;71:444–457. - PubMed
-
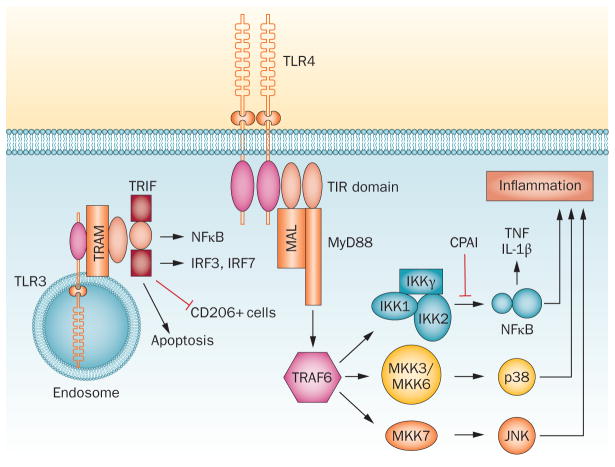
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- NS033997/NS/NINDS NIH HHS/United States
- R01NS44025/NS/NINDS NIH HHS/United States
- R21NS083425/NS/NINDS NIH HHS/United States
- G0802853/MRC_/Medical Research Council/United Kingdom
- R21 NS083425/NS/NINDS NIH HHS/United States
- NS76726/NS/NINDS NIH HHS/United States
- NS082330/NS/NINDS NIH HHS/United States
- R01 NS076726/NS/NINDS NIH HHS/United States
- P01 NS082330/NS/NINDS NIH HHS/United States
- R01 NS044025/NS/NINDS NIH HHS/United States
- WT094823MA/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- R01 NS033997/NS/NINDS NIH HHS/United States
- R21 NS080015/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
