

The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment
- PMID: 25689462
- PMCID: PMC4331492
- DOI: 10.1371/journal.pone.0116515
The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment
Erratum in
-
Correction: The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment.PLoS One. 2015 Mar 30;10(3):e0123612. doi: 10.1371/journal.pone.0123612. eCollection 2015. PLoS One. 2015. PMID: 25823015 Free PMC article. No abstract available.
Abstract
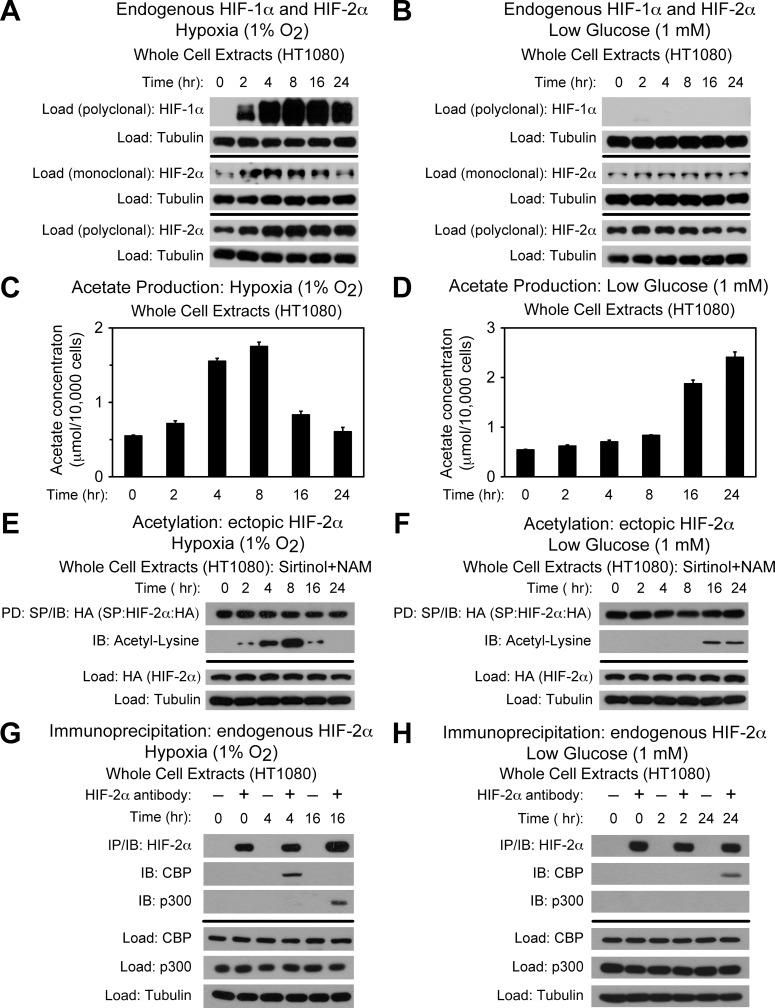
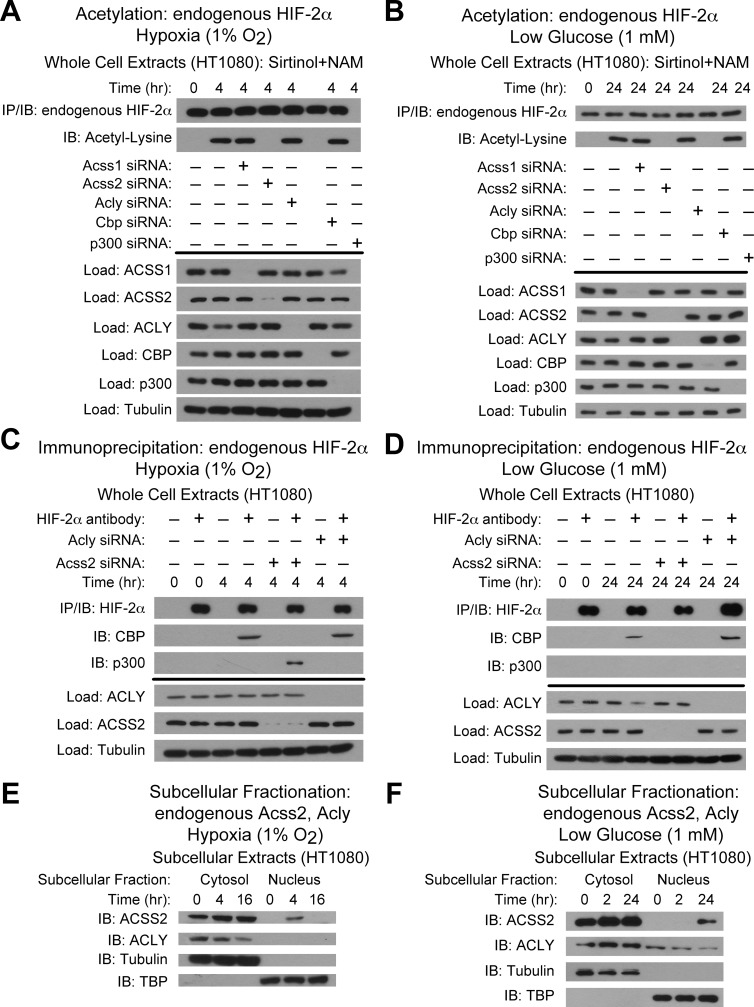
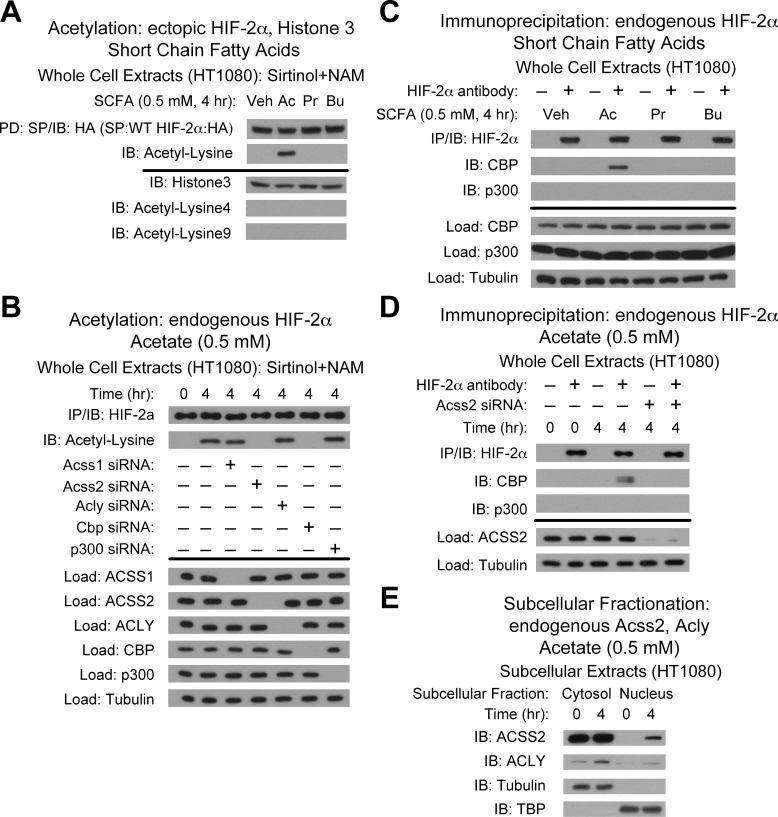
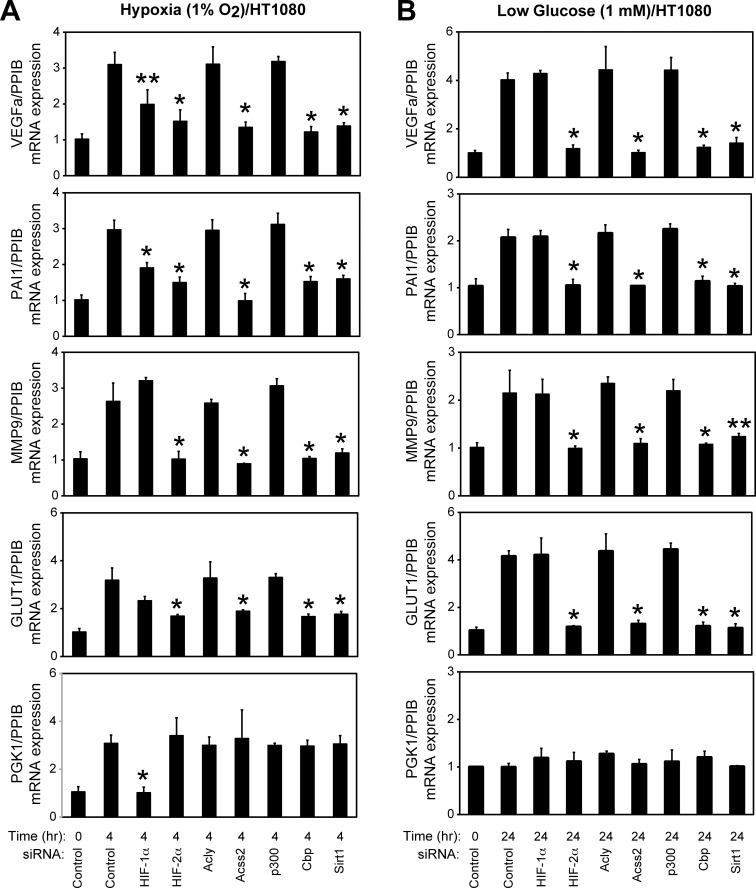
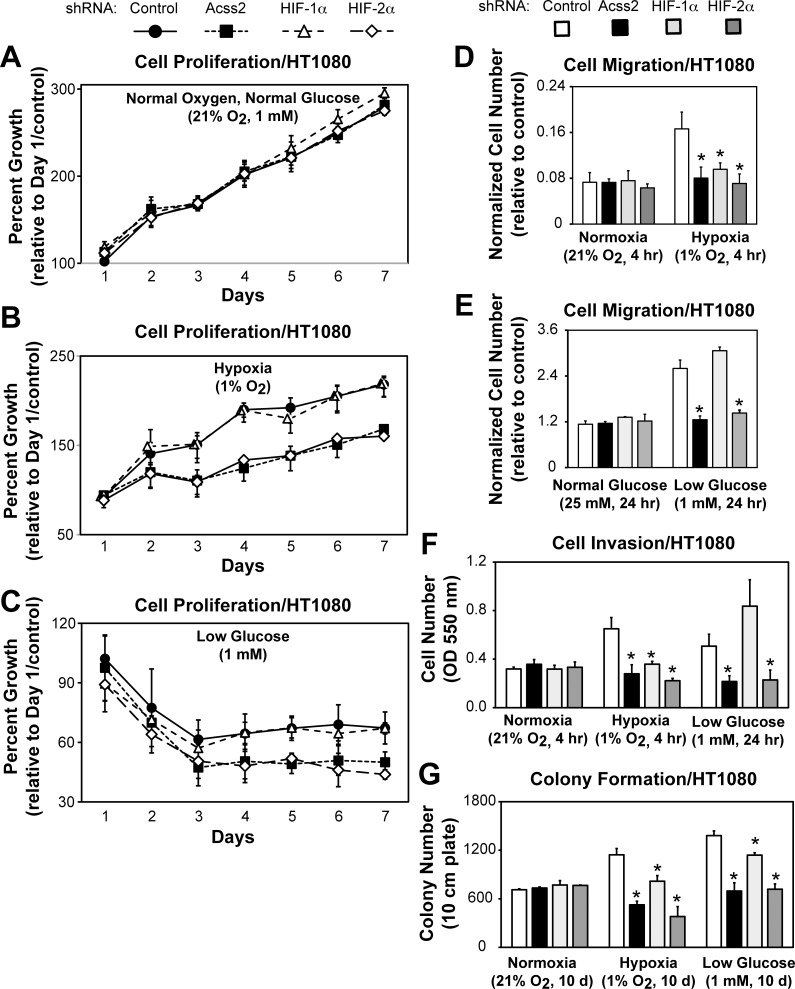
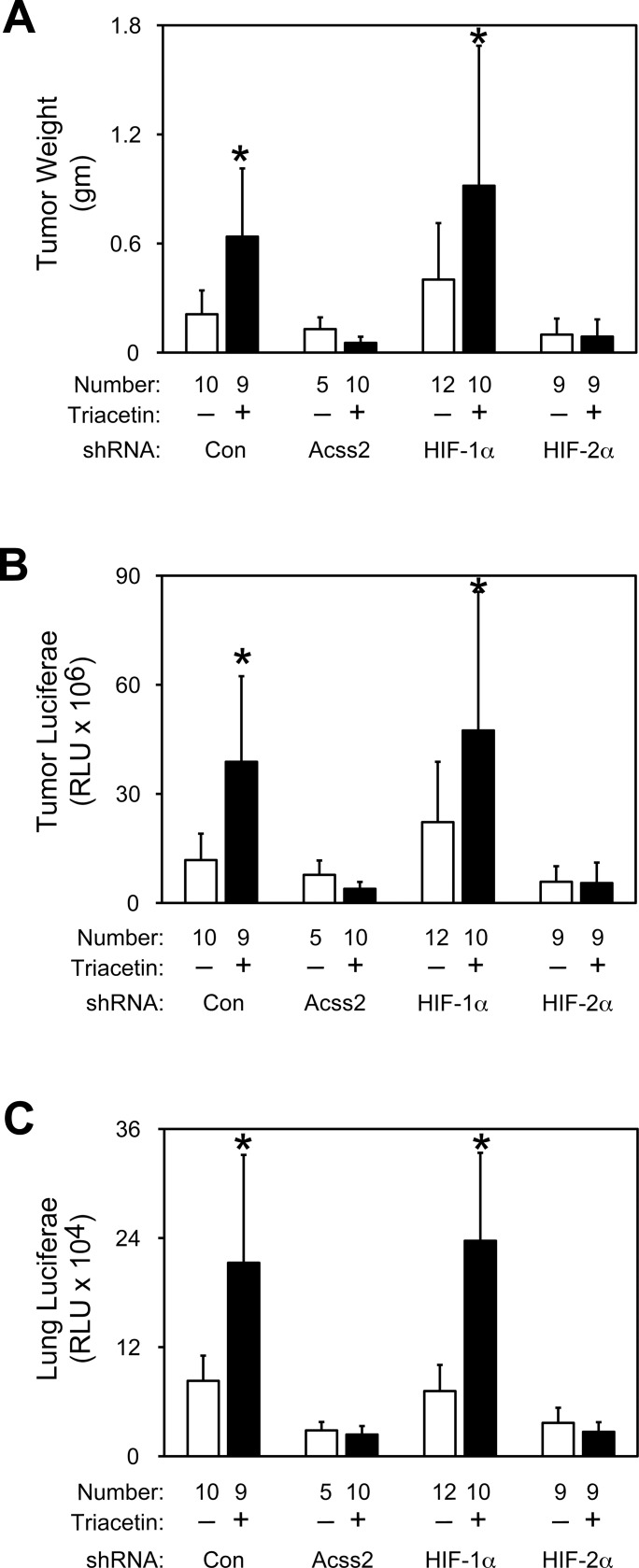
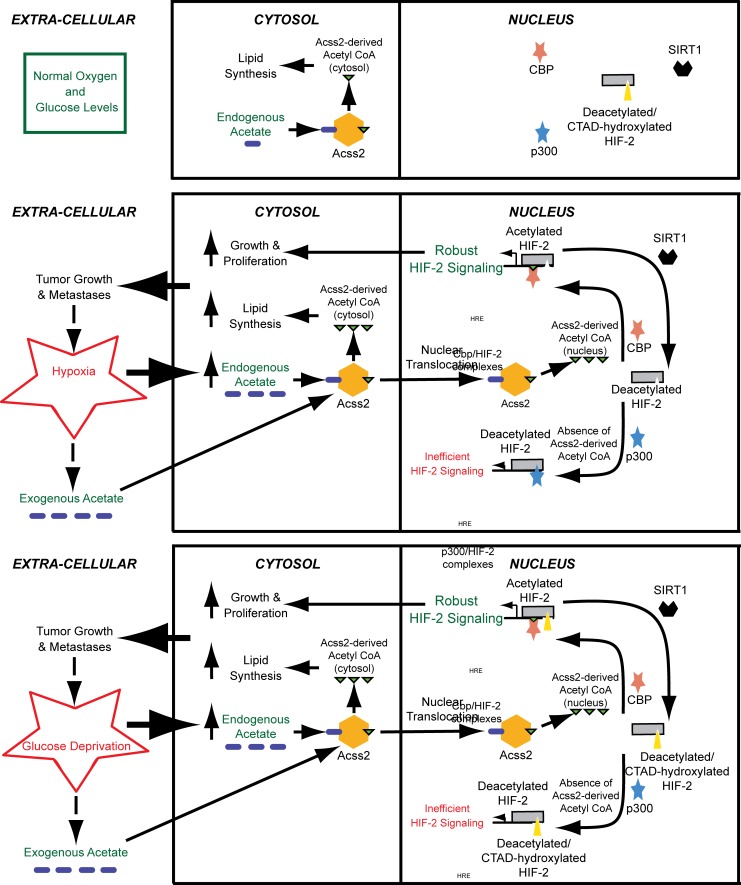
Optimal stress signaling by Hypoxia Inducible Factor 2 (HIF-2) during low oxygen states or hypoxia requires coupled actions of a specific coactivator/lysine acetyltransferase, Creb binding protein (CBP), and a specific deacetylase, Sirtuin 1 (SIRT1). We recently reported that acetylation of HIF-2 by CBP also requires a specific acetyl CoA generator, acetate-dependent acetyl CoA synthetase 2 (ACSS2). In this study, we demonstrate that ACSS2/HIF-2 signaling is active not only during hypoxia, but also during glucose deprivation. Acetate levels increase during stress and coincide with maximal HIF-2α acetylation and CBP/HIF-2α complex formation. Exogenous acetate induces HIF-2α acetylation, CBP/HIF-2α complex formation, and HIF-2 signaling. ACSS2 and HIF-2 are required for maximal colony formation, proliferation, migration, and invasion during stress. Acetate also stimulates flank tumor growth and metastasis in mice in an ACSS2 and HIF-2 dependent manner. Thus, ACSS2/CBP/SIRT1/HIF-2 signaling links nutrient sensing and stress signaling with cancer growth and progression in mammals.
Conflict of interest statement
Figures

Similar articles
-
Coordinate regulation of stress signaling and epigenetic events by Acss2 and HIF-2 in cancer cells.PLoS One. 2017 Dec 27;12(12):e0190241. doi: 10.1371/journal.pone.0190241. eCollection 2017. PLoS One. 2017. PMID: 29281714 Free PMC article.
-
Acss2/HIF-2 signaling facilitates colon cancer growth and metastasis.PLoS One. 2023 Mar 2;18(3):e0282223. doi: 10.1371/journal.pone.0282223. eCollection 2023. PLoS One. 2023. PMID: 36862715 Free PMC article.
-
A substitution mutation in a conserved domain of mammalian acetate-dependent acetyl CoA synthetase 2 results in destabilized protein and impaired HIF-2 signaling.PLoS One. 2019 Nov 14;14(11):e0225105. doi: 10.1371/journal.pone.0225105. eCollection 2019. PLoS One. 2019. PMID: 31725783 Free PMC article.
-
Regulation of gene expression by the hypoxia-inducible factors.Mol Interv. 2002 Jul;2(4):229-43. doi: 10.1124/mi.2.4.229. Mol Interv. 2002. PMID: 14993394 Review.
-
Multiplicity of hypoxia-inducible transcription factors and their connection to the circadian clock in the zebrafish.Physiol Biochem Zool. 2015 Mar-Apr;88(2):146-57. doi: 10.1086/679751. Epub 2015 Jan 14. Physiol Biochem Zool. 2015. PMID: 25730270 Review.
Cited by
-
Correction: The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment.PLoS One. 2015 Mar 30;10(3):e0123612. doi: 10.1371/journal.pone.0123612. eCollection 2015. PLoS One. 2015. PMID: 25823015 Free PMC article. No abstract available.
-
Novel metabolomics-biohumoral biomarkers model for predicting survival of metastatic soft-tissue sarcomas.iScience. 2023 Aug 19;26(10):107678. doi: 10.1016/j.isci.2023.107678. eCollection 2023 Oct 20. iScience. 2023. PMID: 37752948 Free PMC article.
-
Regulation of chromatin and gene expression by metabolic enzymes and metabolites.Nat Rev Mol Cell Biol. 2018 Sep;19(9):563-578. doi: 10.1038/s41580-018-0029-7. Nat Rev Mol Cell Biol. 2018. PMID: 29930302 Free PMC article. Review.
-
Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics, and Oncogenesis - Part 2: Acetate and ACSS2 in Health and Disease.Front Physiol. 2020 Nov 12;11:580171. doi: 10.3389/fphys.2020.580171. eCollection 2020. Front Physiol. 2020. PMID: 33304273 Free PMC article. Review.
-
Molecular Targets and Small Molecules Modulating Acetyl Coenzyme A in Physiology and Diseases.ACS Pharmacol Transl Sci. 2024 Dec 18;8(1):36-46. doi: 10.1021/acsptsci.4c00476. eCollection 2025 Jan 10. ACS Pharmacol Transl Sci. 2024. PMID: 39816789 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
