

On human disease-causing amino acid variants: statistical study of sequence and structural patterns
- PMID: 25689729
- PMCID: PMC4409542
- DOI: 10.1002/humu.22770
On human disease-causing amino acid variants: statistical study of sequence and structural patterns
Abstract
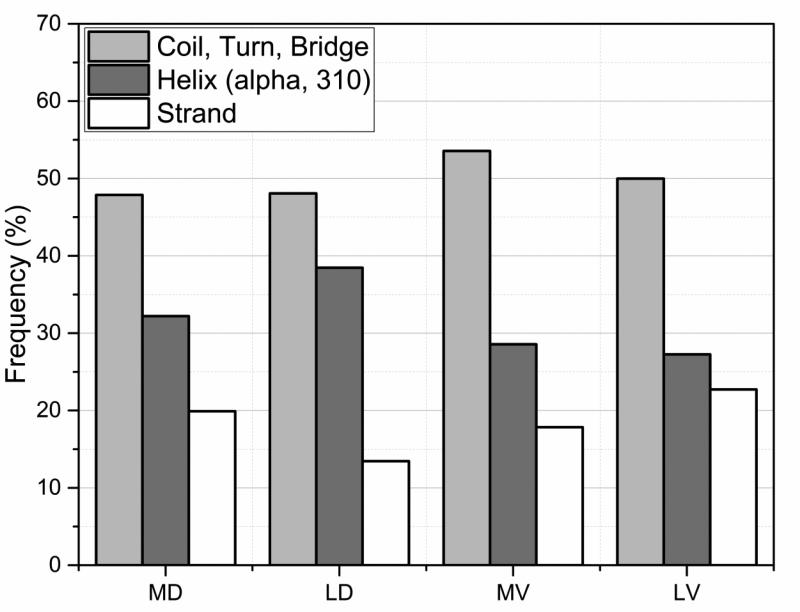
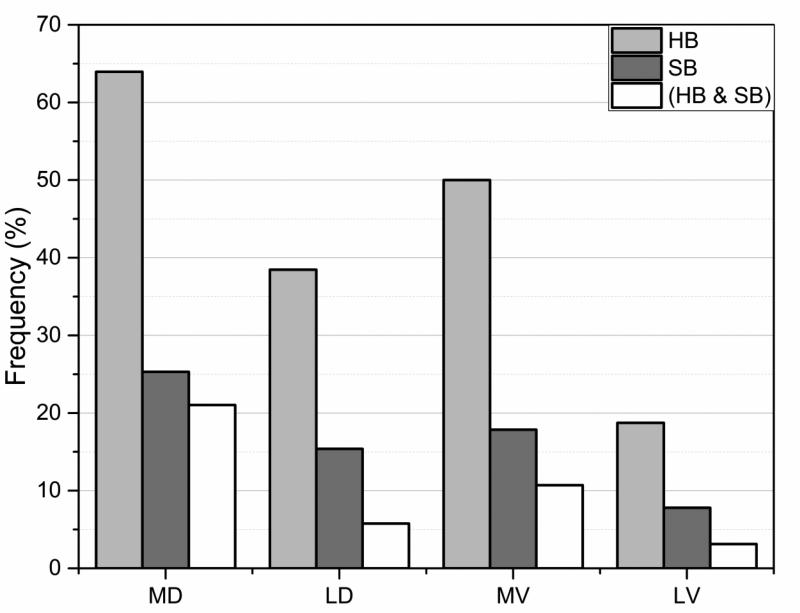
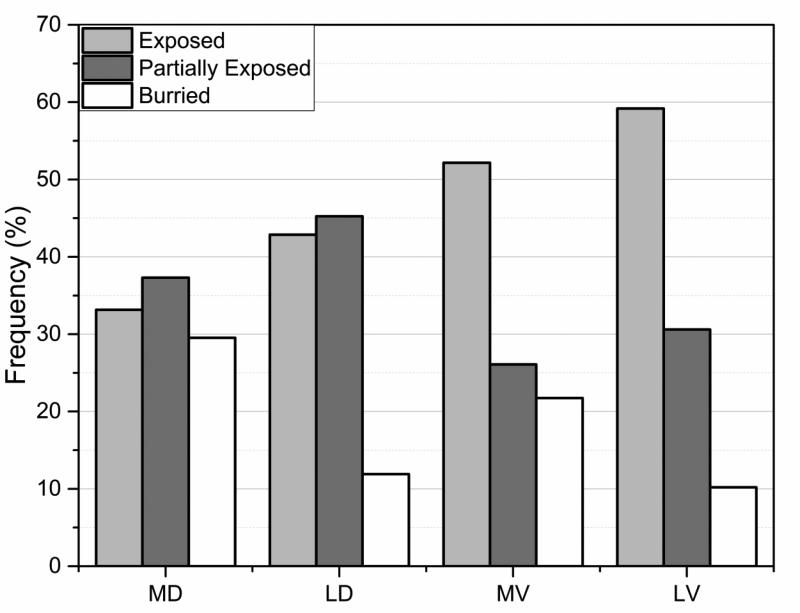
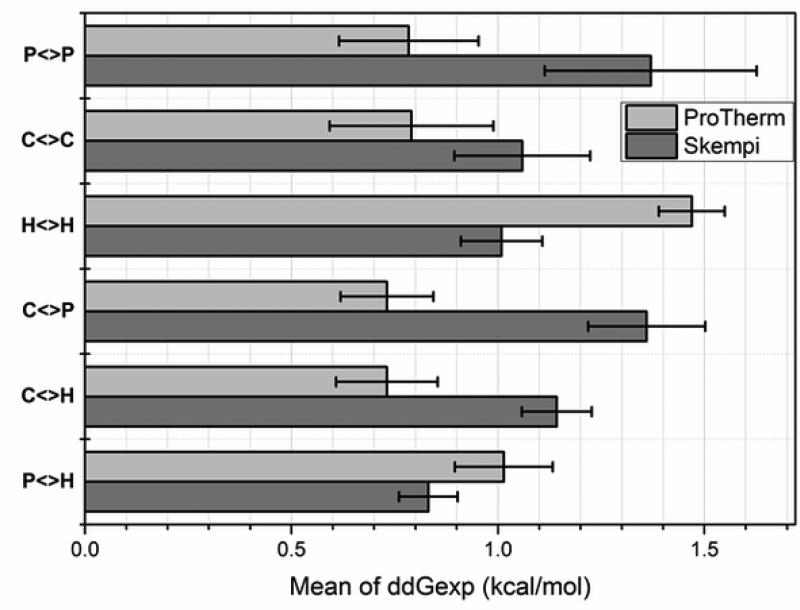
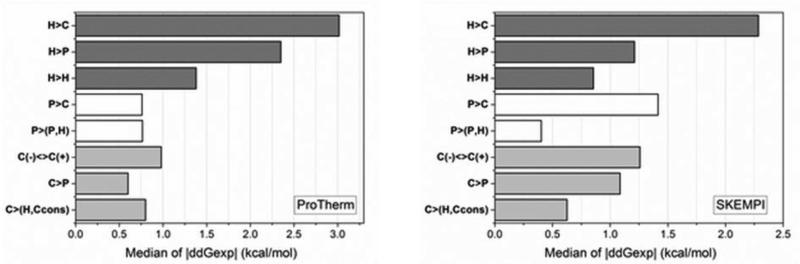
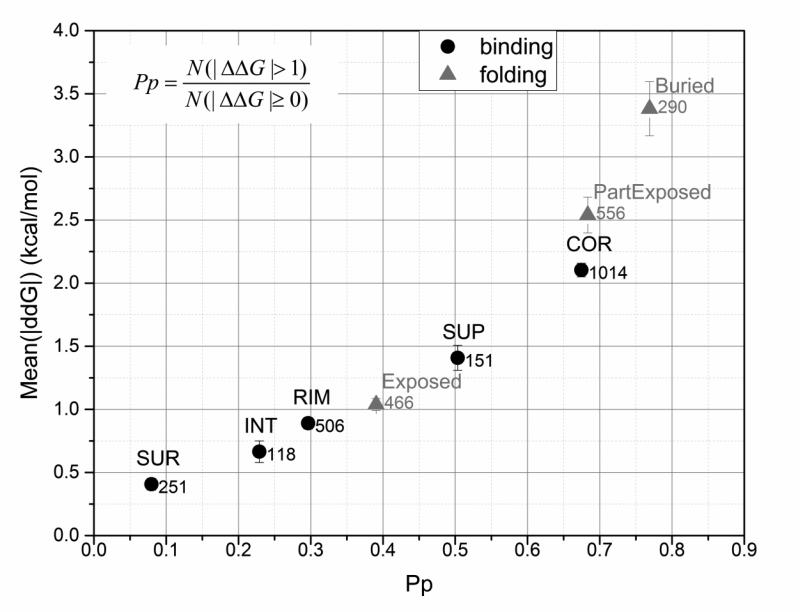
Statistical analysis was carried out on large set of naturally occurring human amino acid variations, and it was demonstrated that there is a preference for some amino acid substitutions to be associated with diseases. At an amino acid sequence level, it was shown that the disease-causing variants frequently involve drastic changes in amino acid physicochemical properties of proteins such as charge, hydrophobicity, and geometry. Structural analysis of variants involved in diseases and being frequently observed in human population showed similar trends: disease-causing variants tend to cause more changes in hydrogen bond network and salt bridges as compared with harmless amino acid mutations. Analysis of thermodynamics data reported in the literature, both experimental and computational, indicated that disease-causing variants tend to destabilize proteins and their interactions, which prompted us to investigate the effects of amino acid mutations on large databases of experimentally measured energy changes in unrelated proteins. Although the experimental datasets were linked neither to diseases nor exclusory to human proteins, the observed trends were the same: amino acid mutations tend to destabilize proteins and their interactions. Having in mind that structural and thermodynamics properties are interrelated, it is pointed out that any large change in any of them is anticipated to cause a disease.
Keywords: amino acid variations; disease mutations; hydrogen bond; structure.
© 2015 WILEY PERIODICALS, INC.
Figures
References
-
- Alexov E, Sternberg M. Understanding molecular effects of naturally occurring genetic differences. J Mol Biol. 2013;425(21):3911–3. - PubMed
-
- Atipairin A, Canyuk B, Ratanaphan A. Substitution of aspartic acid with glutamic acid at position 67 of the BRCA1 RING domain retains ubiquitin ligase activity and zinc (II) binding with a reduced transition temperature. JBIC Journal of Biological Inorganic Chemistry. 2011;16(2):217–226. - PubMed
-
- Baxa U, Steinbacher S, Weintraub A, Huber R, Seckler R. Mutations improving the folding of phage P22 tailspike protein affect its receptor binding activity. J Mol Biol. 1999;293(3):693–701. - PubMed
-
- Benedix A, Becker CM, de Groot BL, Caflisch A, Bockmann RA. Predicting free energy changes using structural ensembles. Nat Methods. 2009;6(1):3–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
