

Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis
- PMID: 25689976
- PMCID: PMC4404431
- DOI: 10.1007/s13311-015-0340-3
Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis
Abstract
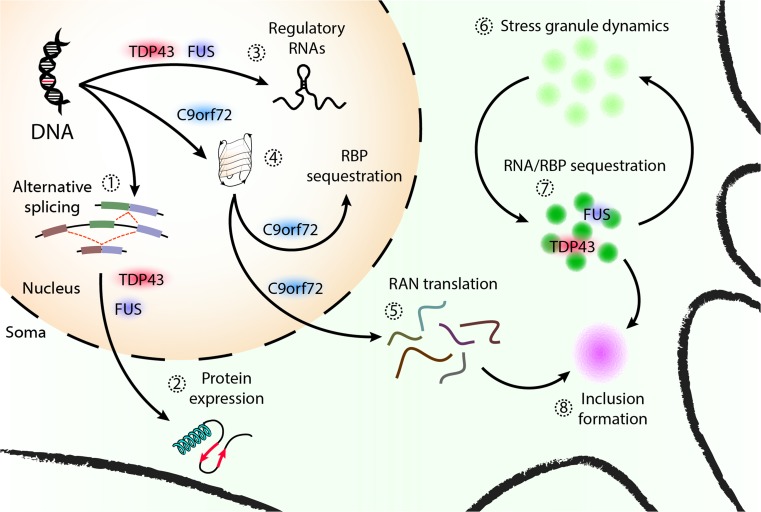
The degeneration of motor neurons in amyotrophic lateral sclerosis (ALS) inevitably causes paralysis and death within a matter of years. Mounting genetic and functional evidence suggest that abnormalities in RNA processing and metabolism underlie motor neuron loss in sporadic and familial ALS. Abnormal localization and aggregation of essential RNA-binding proteins are fundamental pathological features of sporadic ALS, and mutations in genes encoding RNA processing enzymes cause familial disease. Also, expansion mutations occurring in the noncoding region of C9orf72-the most common cause of inherited ALS-result in nuclear RNA foci, underscoring the link between abnormal RNA metabolism and neurodegeneration in ALS. This review summarizes the current understanding of RNA dysfunction in ALS, and builds upon this knowledge base to identify converging mechanisms of neurodegeneration in ALS. Potential targets for therapy development are highlighted, with particular emphasis on early and conserved pathways that lead to motor neuron loss in ALS.
Figures
References
-
- Charcot JM, Joffory A. Deux cas d'atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch Physiol Neurol Pathol. 1869;2:744–754.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
