

Induced pluripotent stem cells model personalized variations in liver disease resulting from α1-antitrypsin deficiency
- PMID: 25690322
- PMCID: PMC4482790
- DOI: 10.1002/hep.27753
Induced pluripotent stem cells model personalized variations in liver disease resulting from α1-antitrypsin deficiency
Abstract
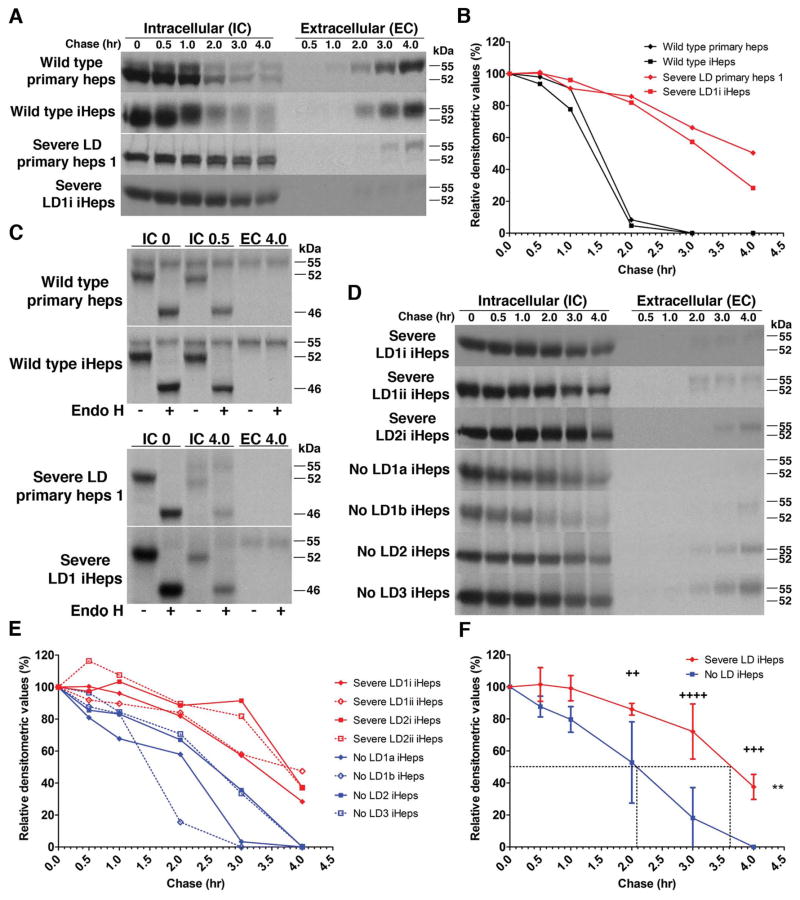
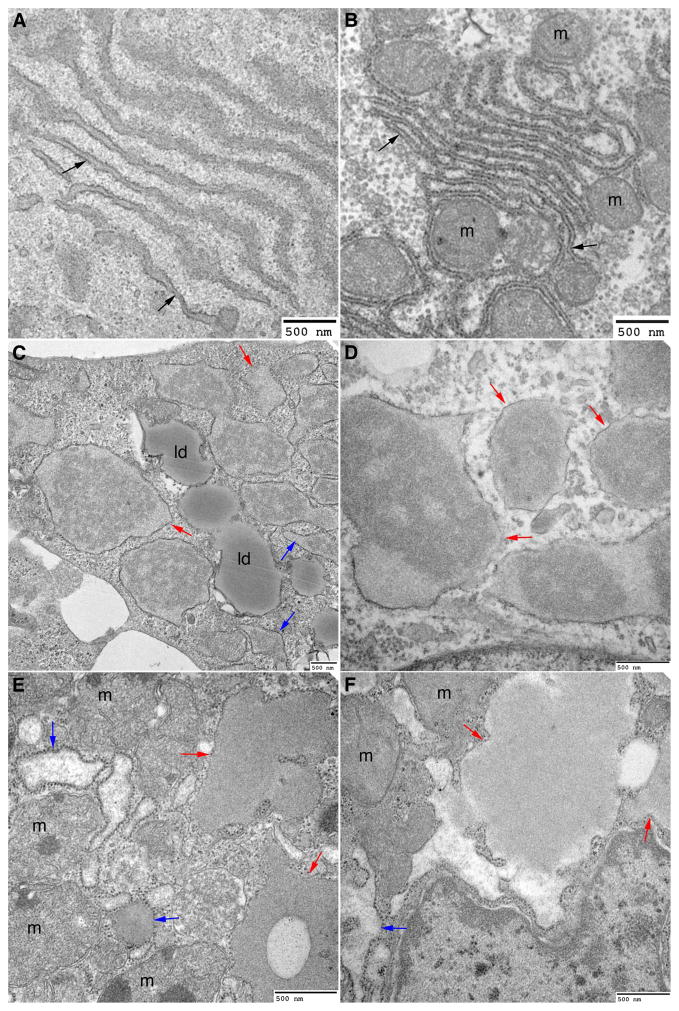
In the classical form of α1-antitrypsin deficiency (ATD), aberrant intracellular accumulation of misfolded mutant α1-antitrypsin Z (ATZ) in hepatocytes causes hepatic damage by a gain-of-function, "proteotoxic" mechanism. Whereas some ATD patients develop severe liver disease (SLD) that necessitates liver transplantation, others with the same genetic defect completely escape this clinical phenotype. We investigated whether induced pluripotent stem cells (iPSCs) from ATD individuals with or without SLD could model these personalized variations in hepatic disease phenotypes. Patient-specific iPSCs were generated from ATD patients and a control and differentiated into hepatocyte-like cells (iHeps) having many characteristics of hepatocytes. Pulse-chase and endoglycosidase H analysis demonstrate that the iHeps recapitulate the abnormal accumulation and processing of the ATZ molecule, compared to the wild-type AT molecule. Measurements of the fate of intracellular ATZ show a marked delay in the rate of ATZ degradation in iHeps from SLD patients, compared to those from no liver disease patients. Transmission electron microscopy showed dilated rough endoplasmic reticulum in iHeps from all individuals with ATD, not in controls, but globular inclusions that are partially covered with ribosomes were observed only in iHeps from individuals with SLD.
Conclusion: iHeps model the individual disease phenotypes of ATD patients with more rapid degradation of misfolded ATZ and lack of globular inclusions in cells from patients who have escaped liver disease. The results support the concept that "proteostasis" mechanisms, such as intracellular degradation pathways, play a role in observed variations in clinical phenotype and show that iPSCs can potentially be used to facilitate predictions of disease susceptibility for more precise and timely application of therapeutic strategies.
© 2015 by the American Association for the Study of Liver Diseases.
Conflict of interest statement
Potential conflict of interest: Nothing to report.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials