

E6^E7, a novel splice isoform protein of human papillomavirus 16, stabilizes viral E6 and E7 oncoproteins via HSP90 and GRP78
- PMID: 25691589
- PMCID: PMC4337564
- DOI: 10.1128/mBio.02068-14
E6^E7, a novel splice isoform protein of human papillomavirus 16, stabilizes viral E6 and E7 oncoproteins via HSP90 and GRP78
Abstract
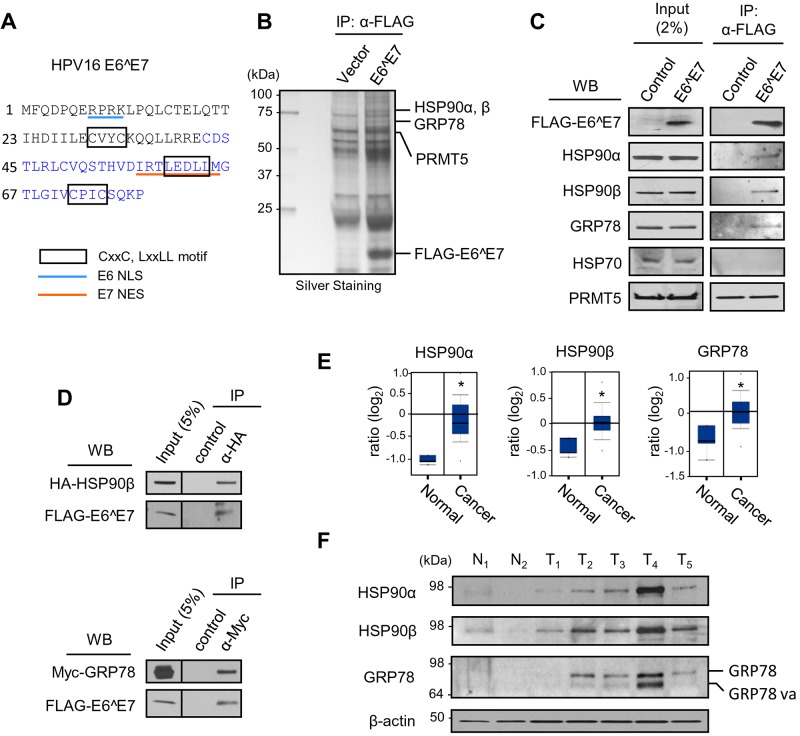
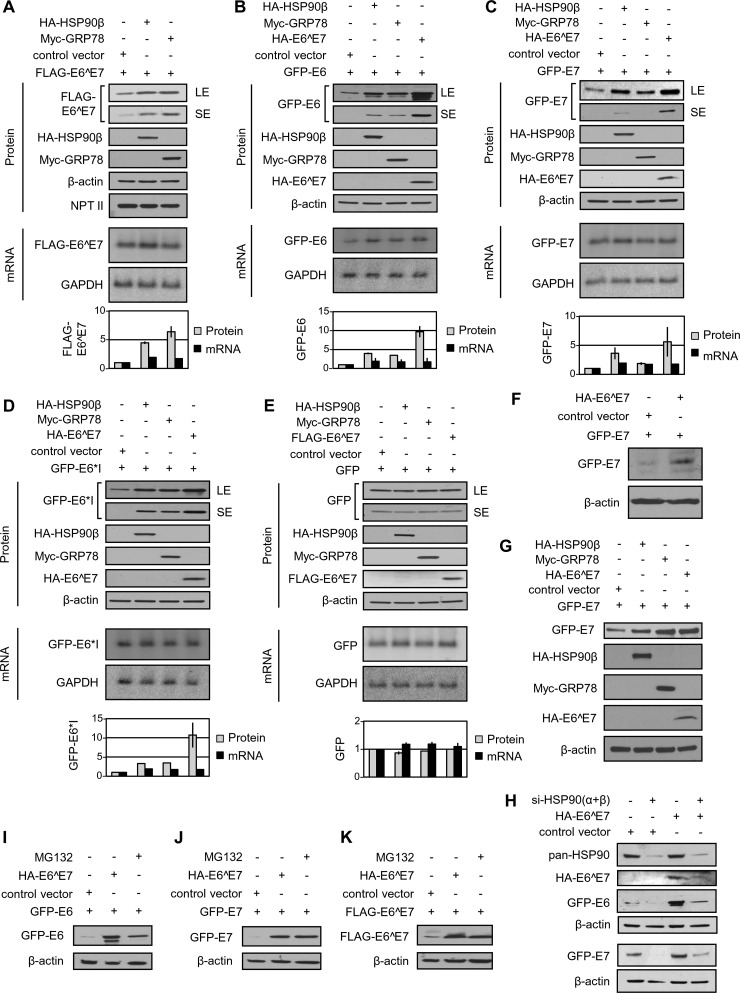
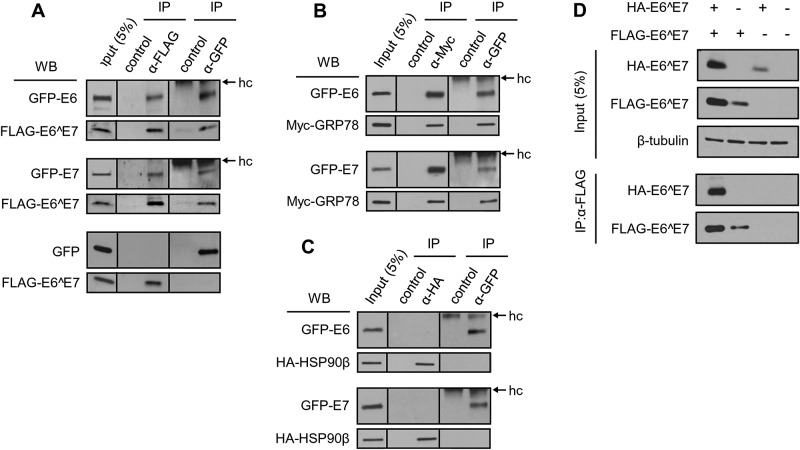
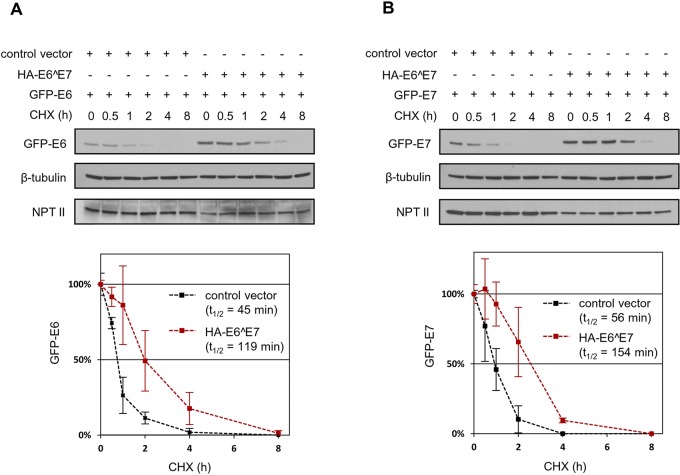
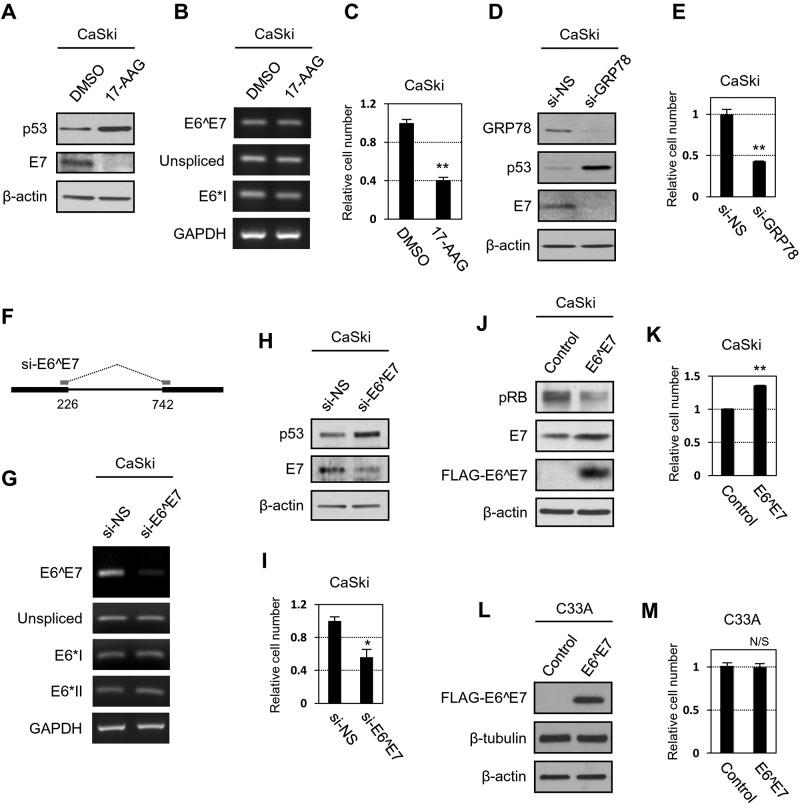
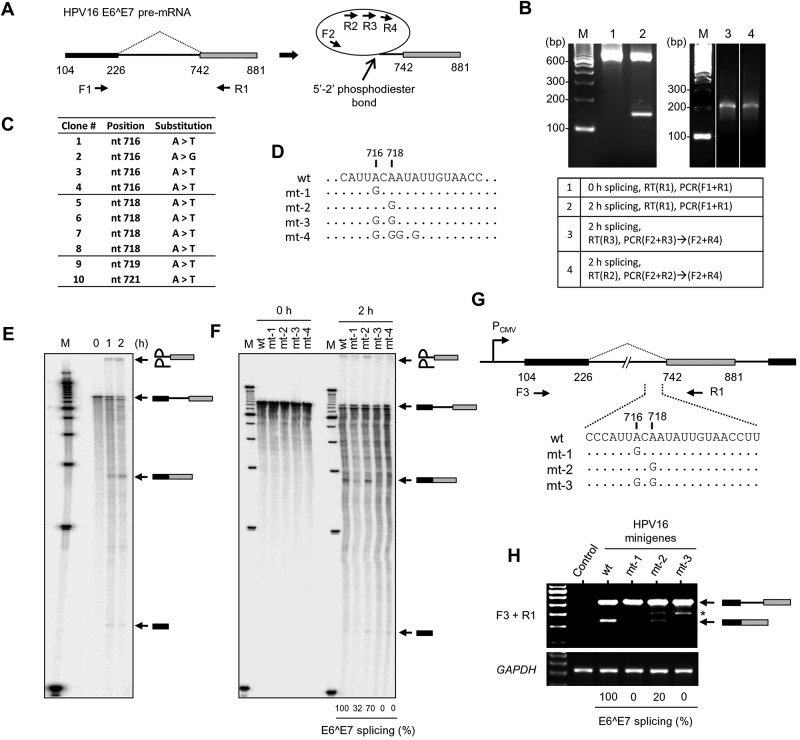
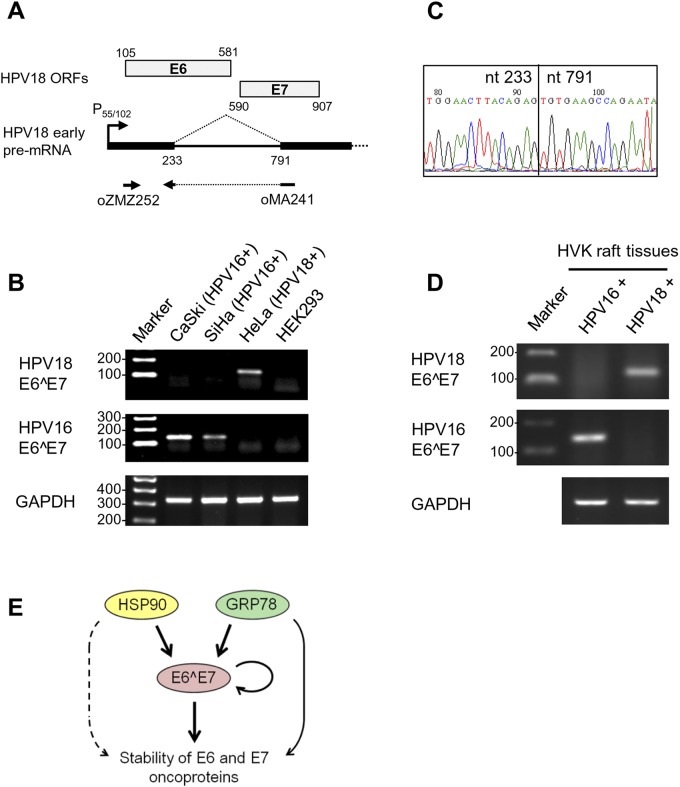
Transcripts of human papillomavirus 16 (HPV16) E6 and E7 oncogenes undergo alternative RNA splicing to produce multiple splice isoforms. However, the importance of these splice isoforms is poorly understood. Here we report a critical role of E6^E7, a novel isoform containing the 41 N-terminal amino acid (aa) residues of E6 and the 38 C-terminal aa residues of E7, in the regulation of E6 and E7 stability. Through mass spectrometric analysis, we identified that HSP90 and GRP78, which are frequently upregulated in cervical cancer tissues, are two E6^E7-interacting proteins responsible for the stability and function of E6^E7, E6, and E7. Although GRP78 and HSP90 do not bind each other, GRP78, but not HSP90, interacts with E6 and E7. E6^E7 protein, in addition to self-binding, interacts with E6 and E7 in the presence of GRP78 and HSP90, leading to the stabilization of E6 and E7 by prolonging the half-life of each protein. Knocking down E6^E7 expression in HPV16-positive CaSki cells by a splice junction-specific small interfering RNA (siRNA) destabilizes E6 and E7 and prevents cell growth. The same is true for the cells with a GRP78 knockdown or in the presence of an HSP90 inhibitor. Moreover, mapping and alignment analyses for splicing elements in 36 alpha-HPVs (α-HPVs) suggest the possible expression of E6^E7 mostly by other oncogenic or possibly oncogenic α-HPVs (HPV18, -30, -31, -39, -42, -45, -56, -59, -70, and -73). HPV18 E6^E7 is detectable in HPV18-positive HeLa cells and HPV18-infected raft tissues. All together, our data indicate that viral E6^E7 and cellular GRP78 or HSP90 might be novel targets for cervical cancer therapy.
Importance: HPV16 is the most prevalent HPV genotype, being responsible for 60% of invasive cervical cancer cases worldwide. What makes HPV16 so potent in the development of cervical cancer remains a mystery. We discovered in this study that, besides producing two well-known oncoproteins, E6 and E7, seen in other high-risk HPVs, HPV16 produces E6^E7, a novel splice isoform of E6 and E7. E6^E7, in addition to self-interacting, binds cellular chaperone proteins, HSP90 and GRP78, and viral E6 and E7 to increase the steady-state levels and half-lives of viral oncoproteins, leading to cell proliferation. The splicing cis elements in the regulation of HPV16 E6^E7 production are highly conserved in 11 oncogenic or possibly oncogenic HPVs, and we confirmed the production of HPV18 E6^E7 in HPV18-infected cells. This study provides new insight into the mechanism of splicing, the interplay between different products of the polycistronic viral message, and the role of the host chaperones as they function.
Copyright © 2015 Ajiro and Zheng.
Figures
References
-
- Howley PM, Lowy DR. 2007. Papillomaviruses, p 2299–2354. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
-
- Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, Snijders PJ, Meijer CJ, International Agency for Research on Cancer Multicenter Cervical Cancer Study Group . 2003. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 348:518–527. doi:10.1056/NEJMoa021641. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
