

Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes
- PMID: 25691889
- PMCID: PMC4315098
- DOI: 10.3389/fgene.2015.00003
Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes
Abstract
The human nuclear and mitochondrial genomes co-exist within each cell. While the mitochondrial genome encodes for a limited number of proteins, transfer RNAs, and ribosomal RNAs, the vast majority of mitochondrial proteins are encoded in the nuclear genome. Of the multitude of mitochondrial disorders known to date, only a fifth are maternally inherited. The recent characterization of the mitochondrial proteome therefore serves as an important step toward delineating the nosology of a large spectrum of phenotypically heterogeneous diseases. Following the identification of the first nuclear gene defect to underlie a mitochondrial disorder, a plenitude of genetic variants that provoke mitochondrial pathophysiology have been molecularly elucidated and classified into six categories that impact: (1) oxidative phosphorylation (subunits and assembly factors); (2) mitochondrial DNA maintenance and expression; (3) mitochondrial protein import and assembly; (4) mitochondrial quality control (chaperones and proteases); (5) iron-sulfur cluster homeostasis; and (6) mitochondrial dynamics (fission and fusion). Here, we propose that an additional class of genetic variant be included in the classification schema to acknowledge the role of genetic defects in phospholipid biosynthesis, remodeling, and metabolism in mitochondrial pathophysiology. This seventh class includes a small but notable group of nuclear-encoded proteins whose dysfunction impacts normal mitochondrial phospholipid metabolism. The resulting human disorders present with a diverse array of pathologic consequences that reflect the variety of functions that phospholipids have in mitochondria and highlight the important role of proper membrane homeostasis in mitochondrial biology.
Keywords: Barth syndrome; DCMA; MEGDEL; Sengers syndrome; cardiolipin; hereditary spastic paraplegia; mitochondrial disease; phospholipid metabolism.
Figures
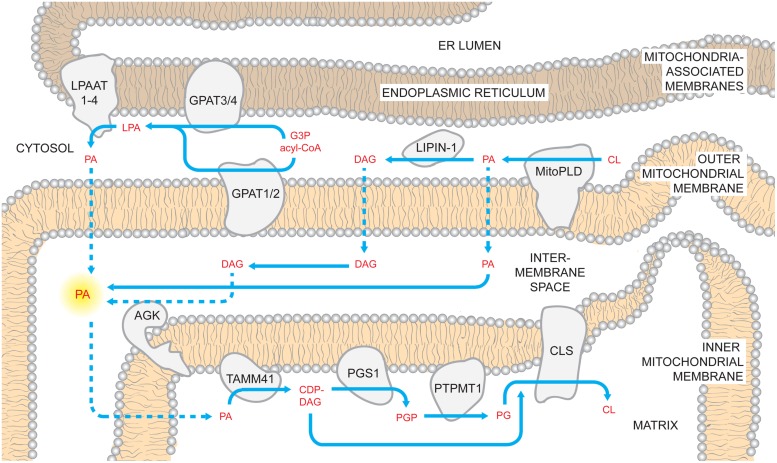
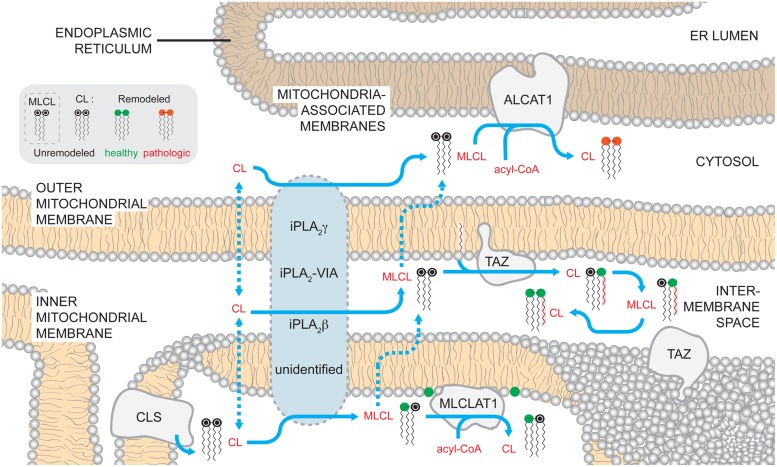
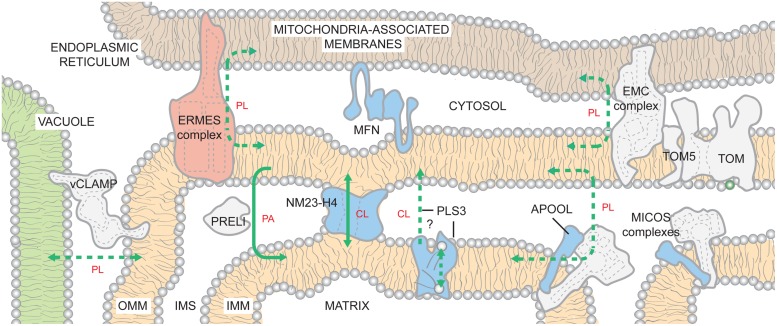
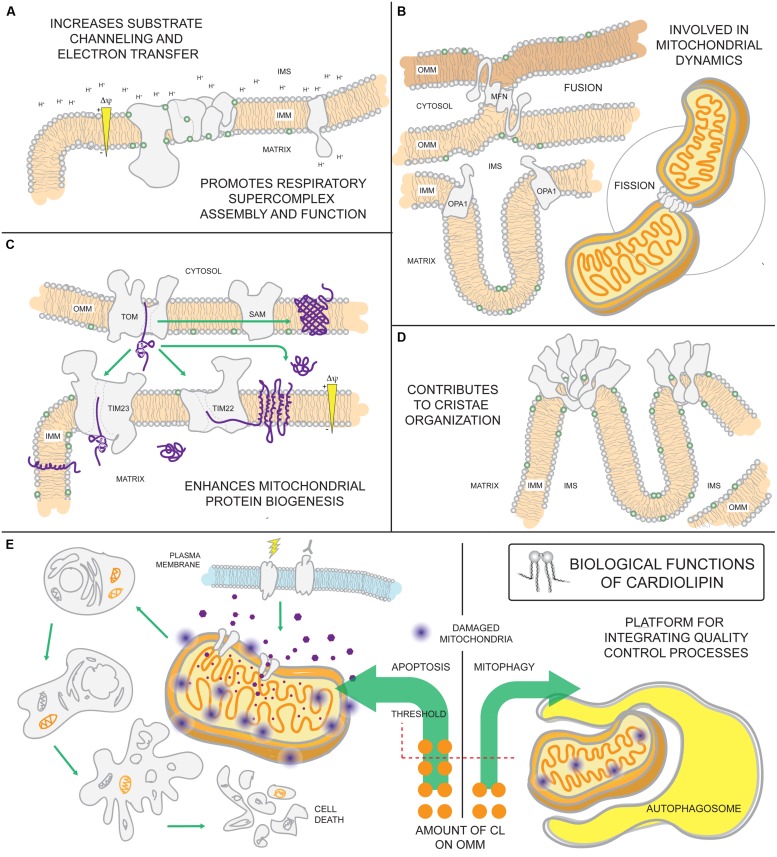
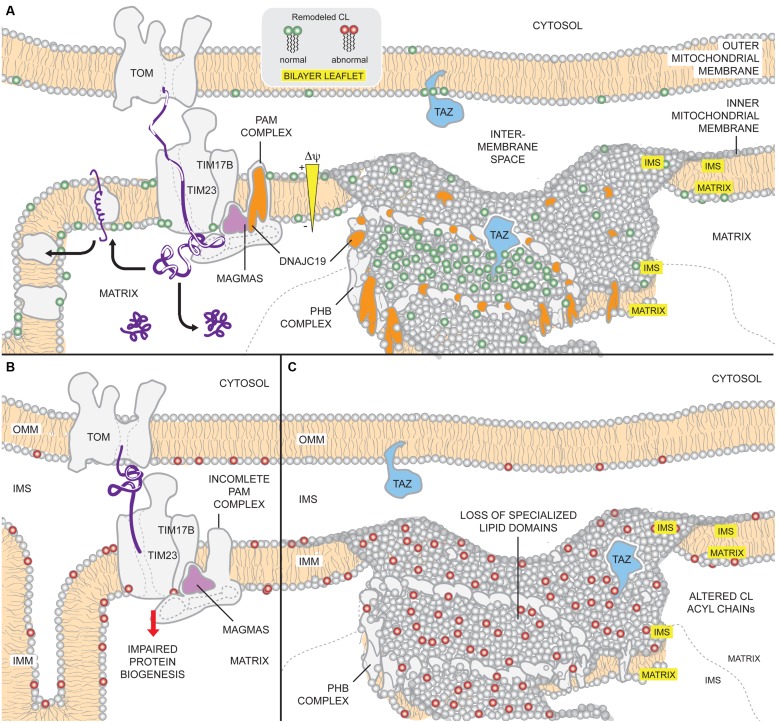
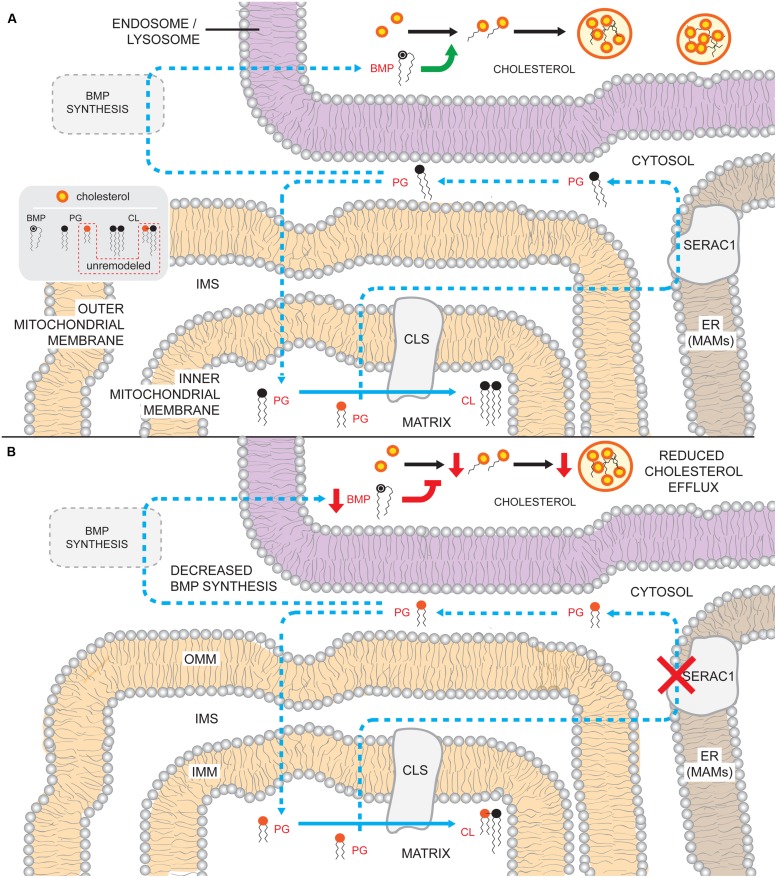
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases