

Transcription factor binding dynamics during human ES cell differentiation
- PMID: 25693565
- PMCID: PMC4499331
- DOI: 10.1038/nature14233
Transcription factor binding dynamics during human ES cell differentiation
Abstract
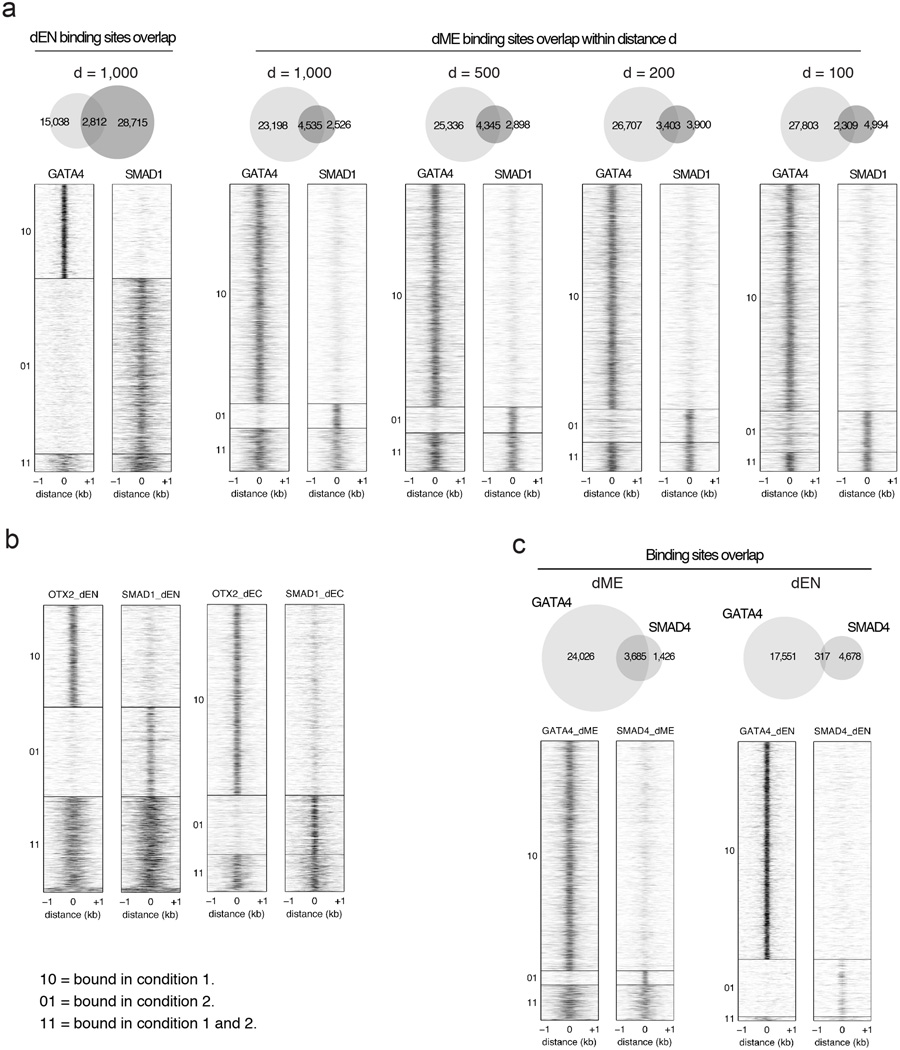
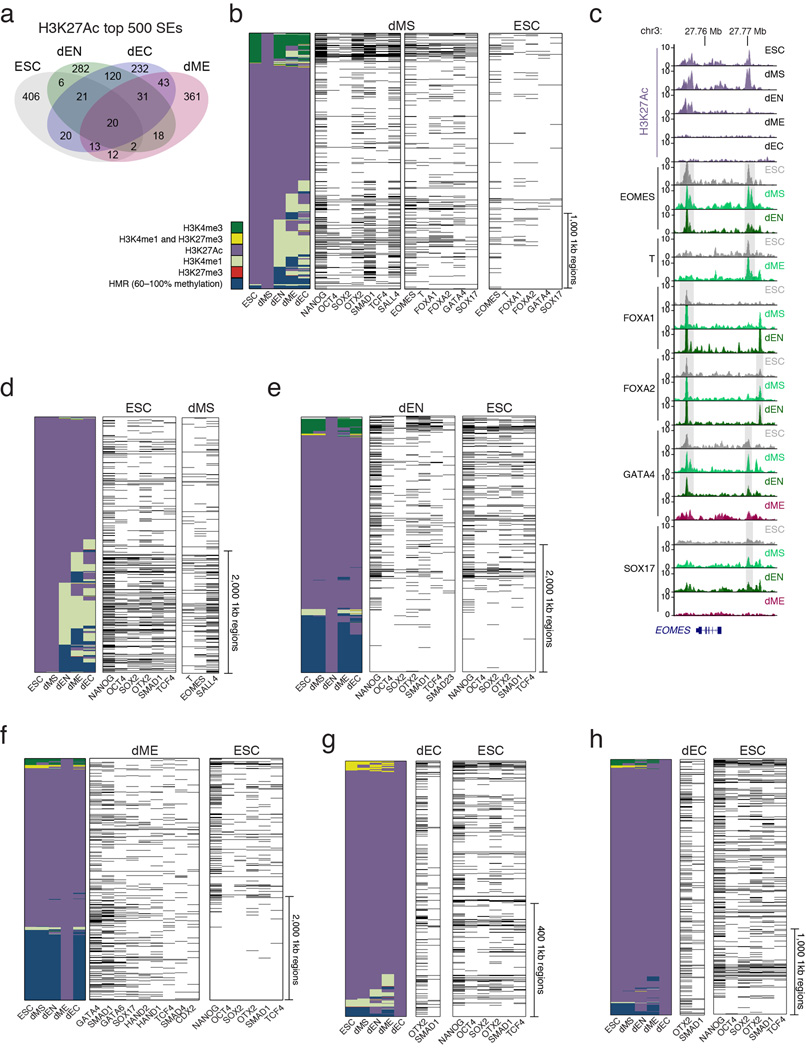
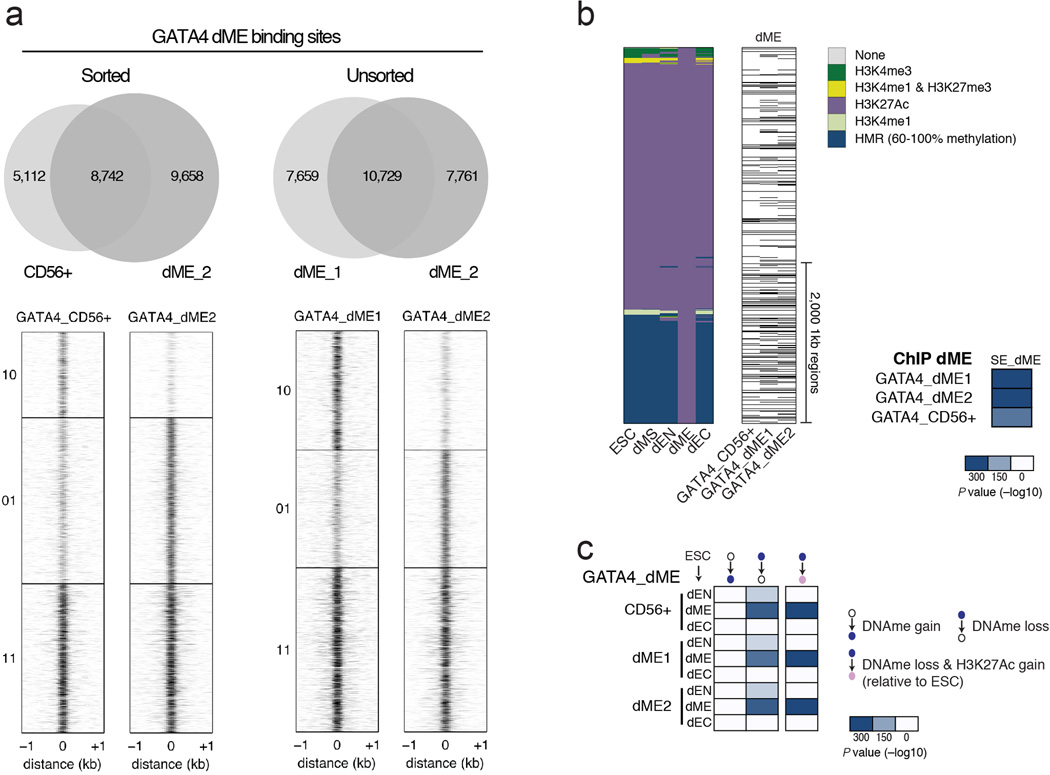
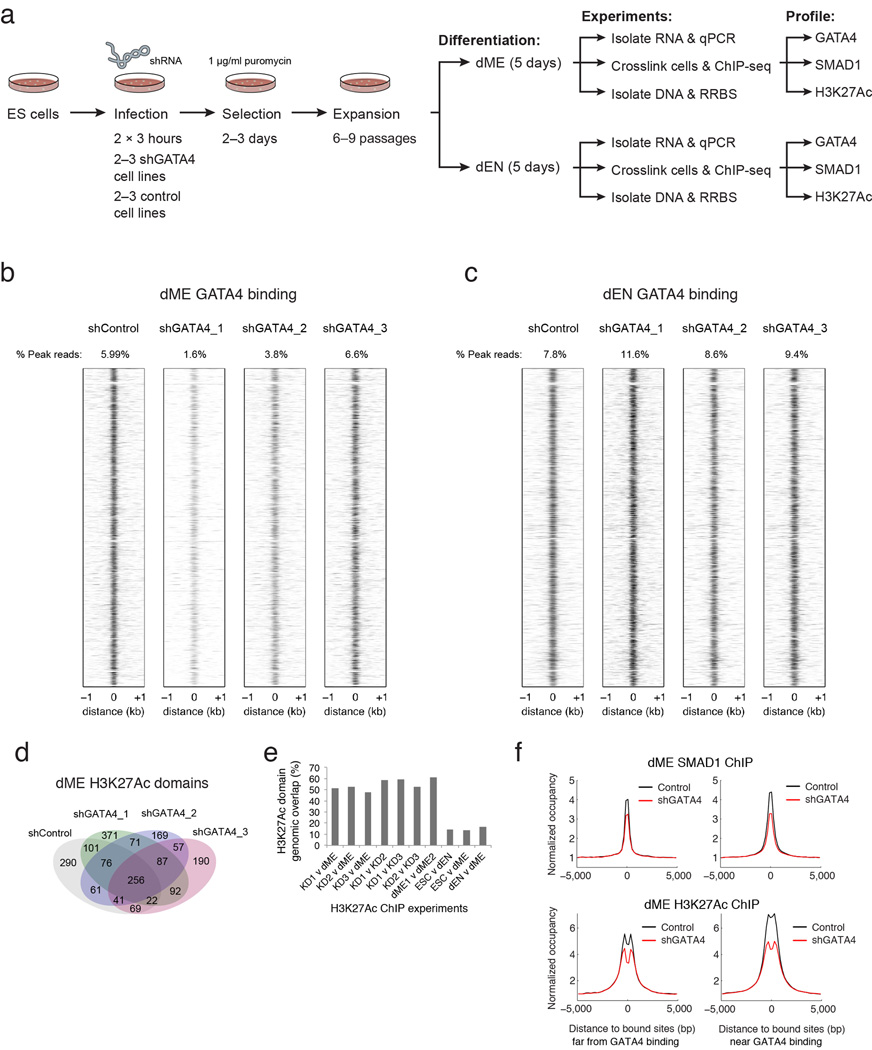
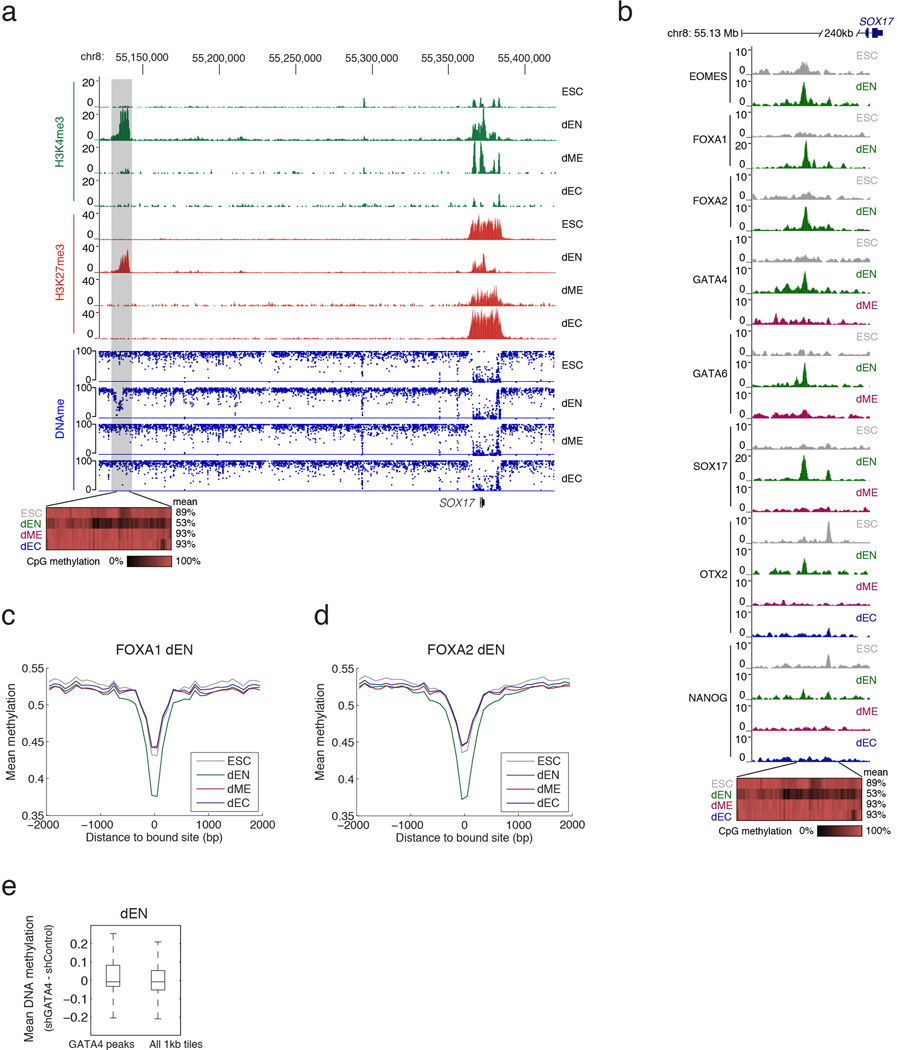
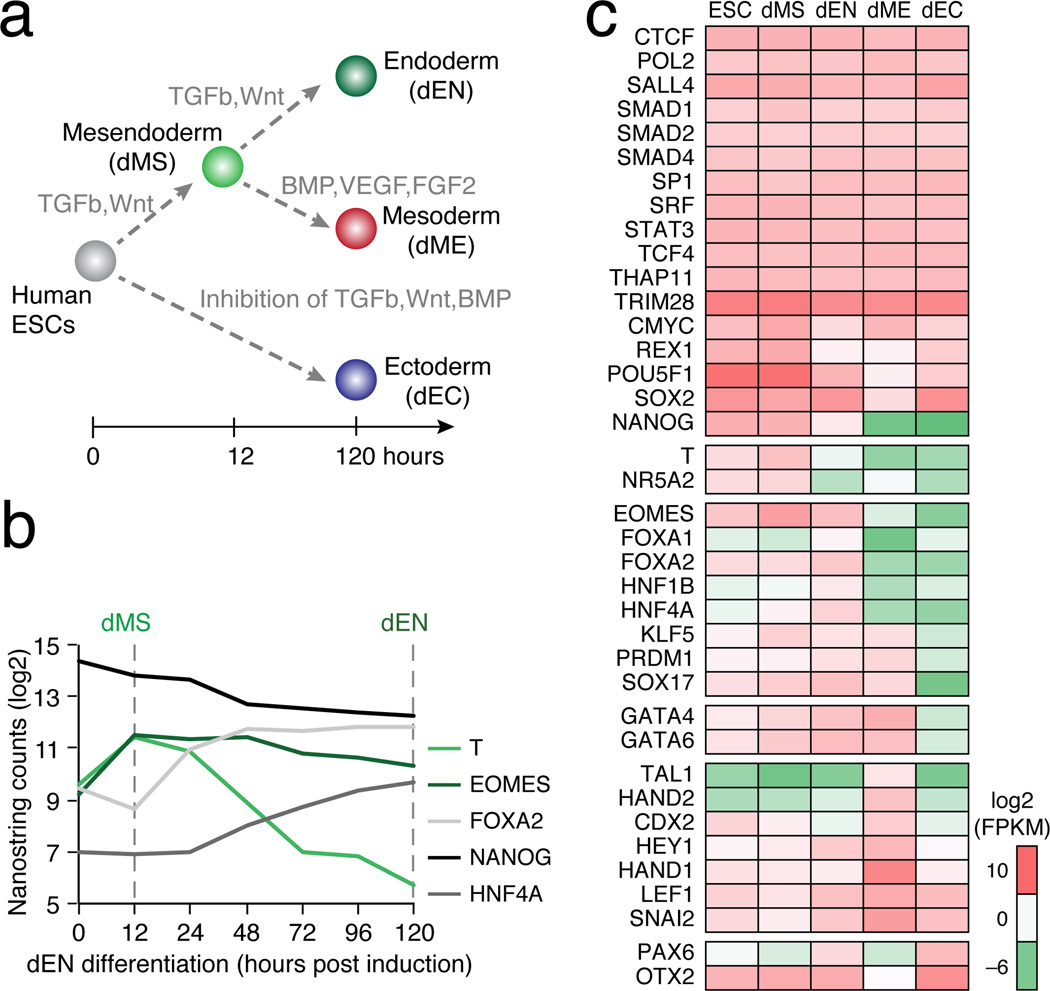
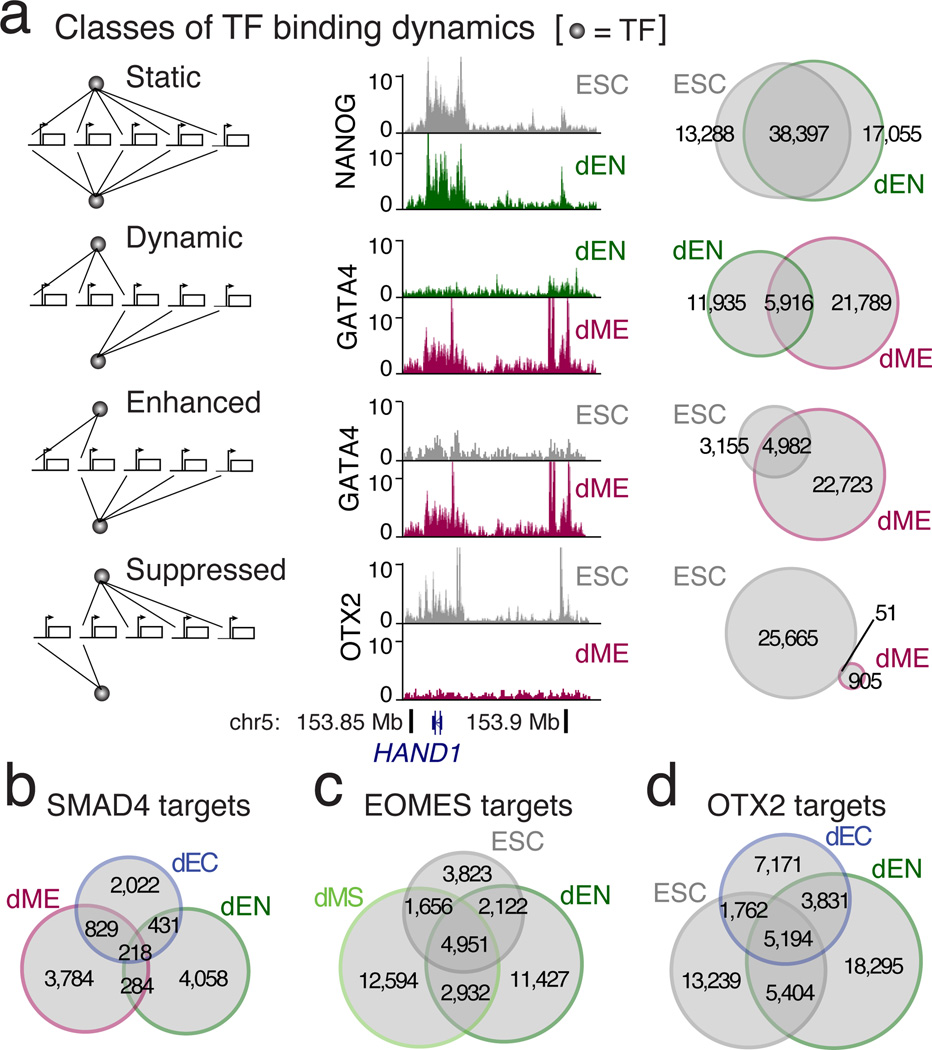
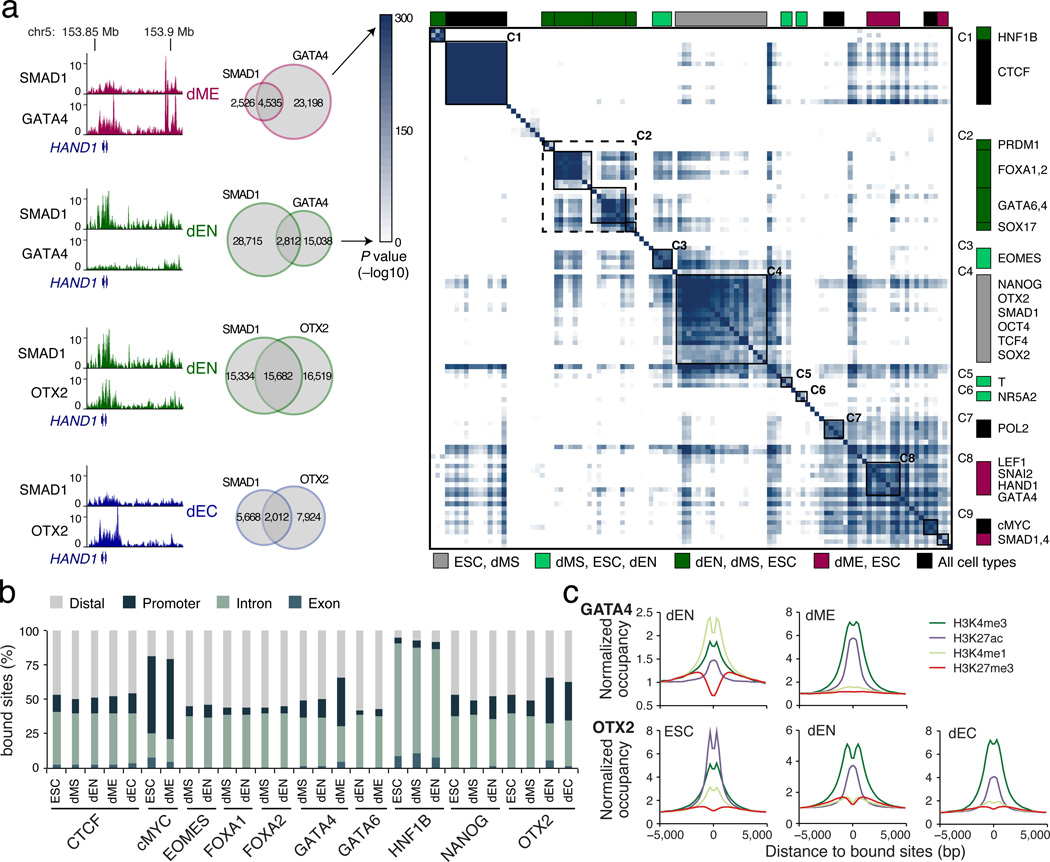
Pluripotent stem cells provide a powerful system to dissect the underlying molecular dynamics that regulate cell fate changes during mammalian development. Here we report the integrative analysis of genome-wide binding data for 38 transcription factors with extensive epigenome and transcriptional data across the differentiation of human embryonic stem cells to the three germ layers. We describe core regulatory dynamics and show the lineage-specific behaviour of selected factors. In addition to the orchestrated remodelling of the chromatin landscape, we find that the binding of several transcription factors is strongly associated with specific loss of DNA methylation in one germ layer, and in many cases a reciprocal gain in the other layers. Taken together, our work shows context-dependent rewiring of transcription factor binding, downstream signalling effectors, and the epigenome during human embryonic stem cell differentiation.
Figures
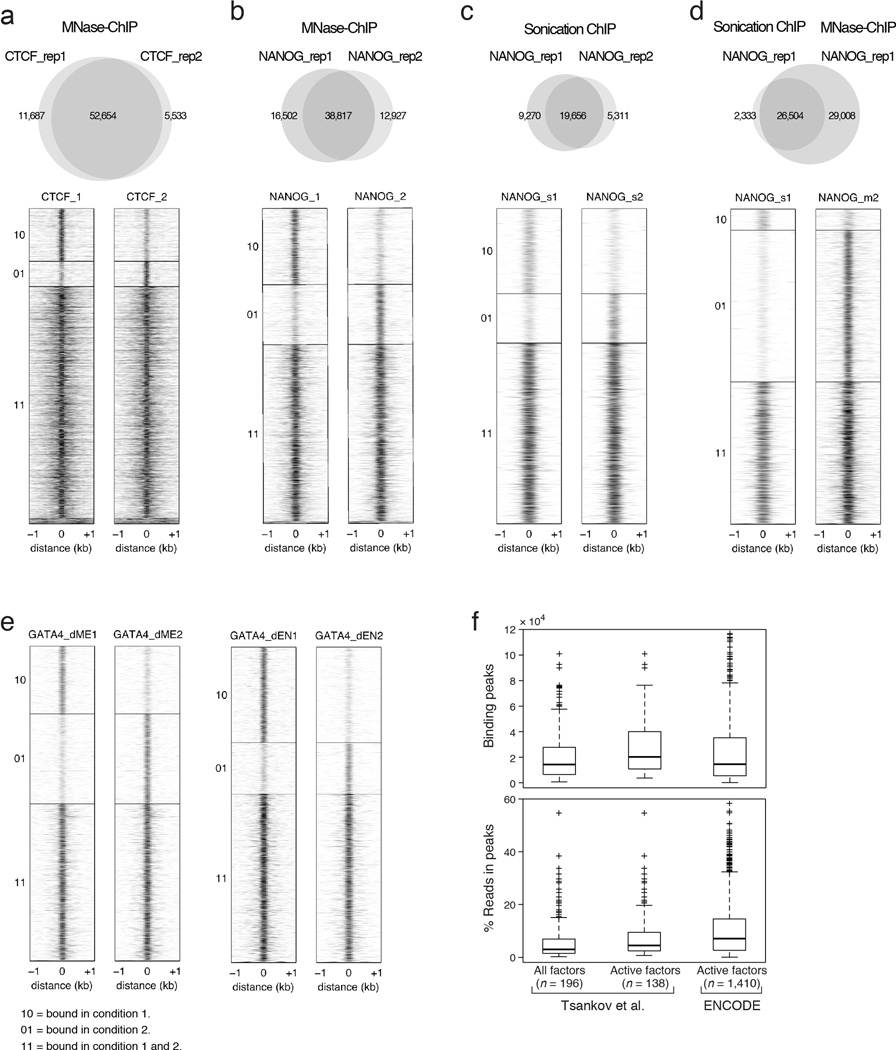
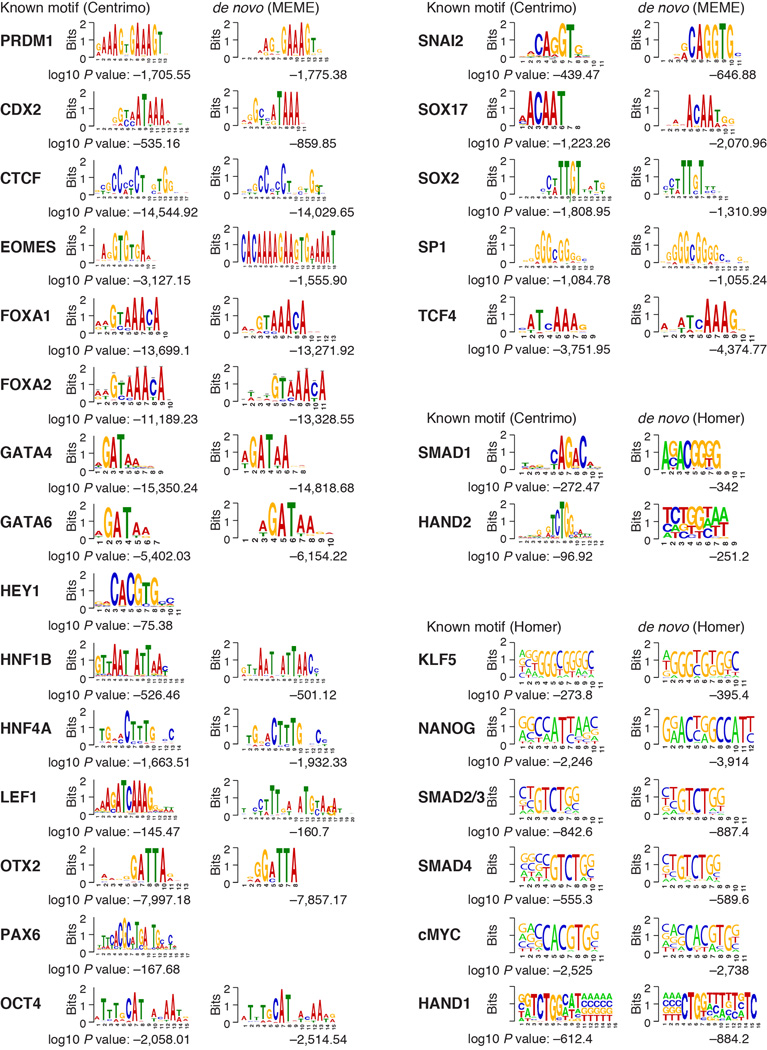
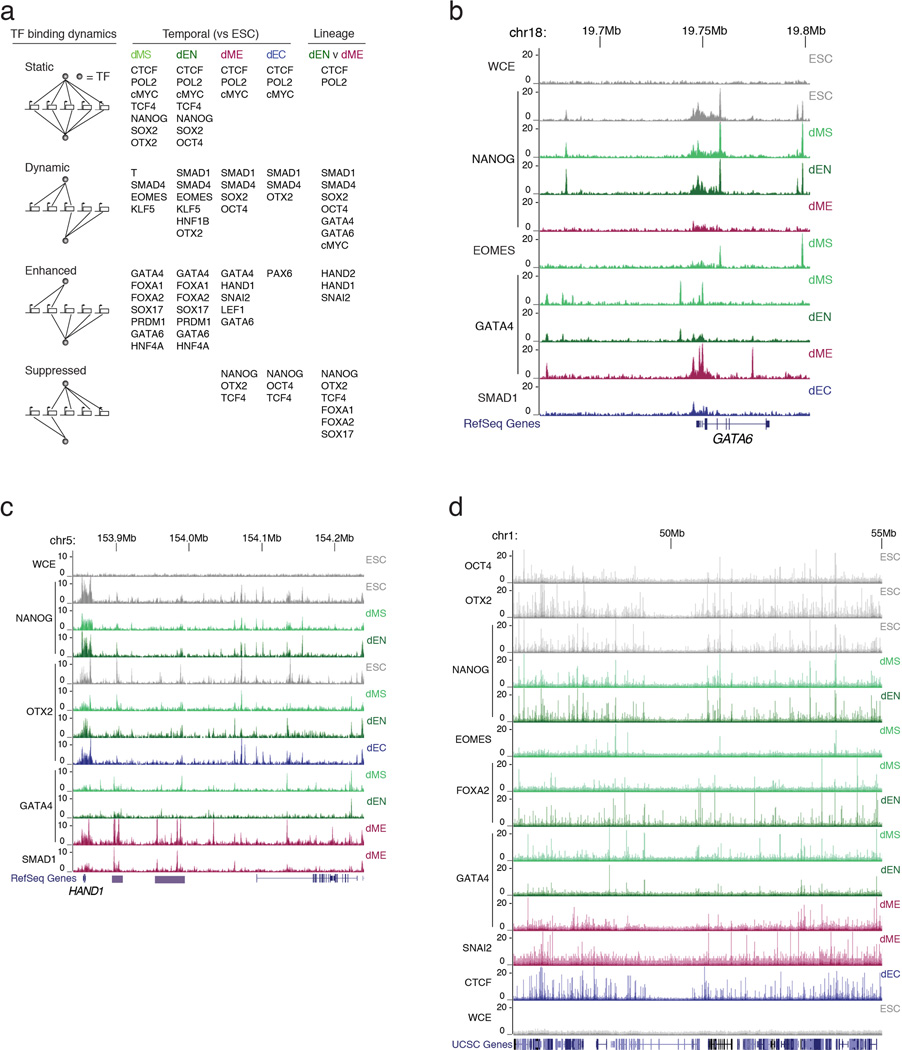
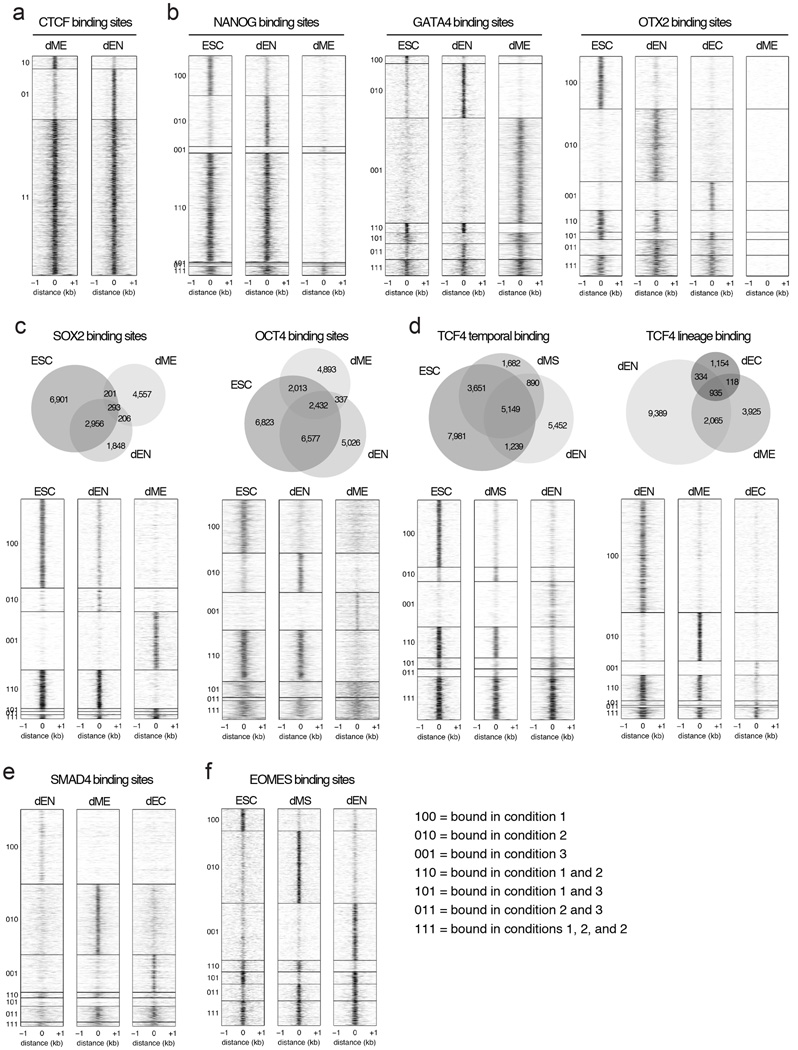

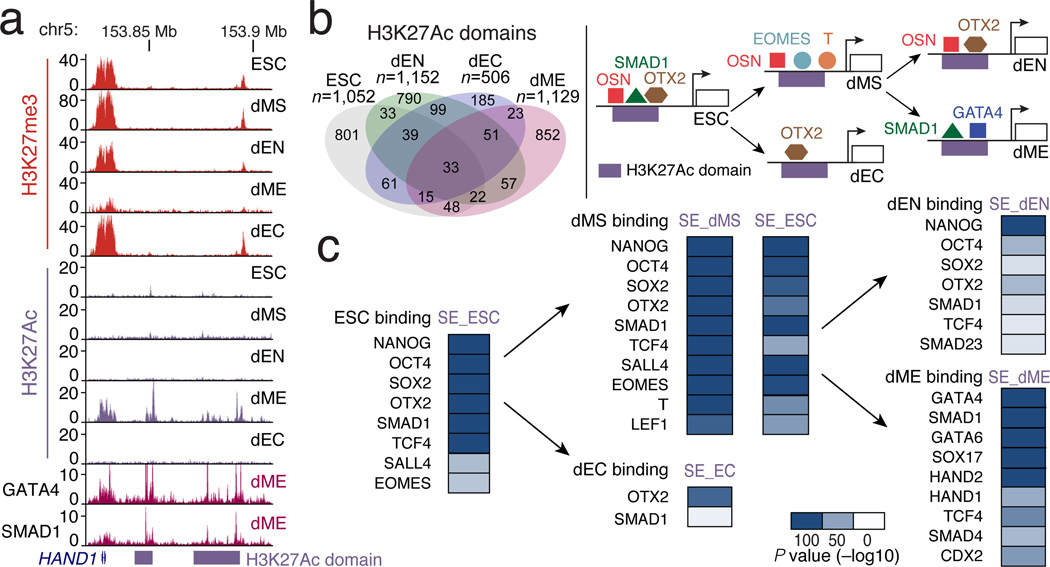
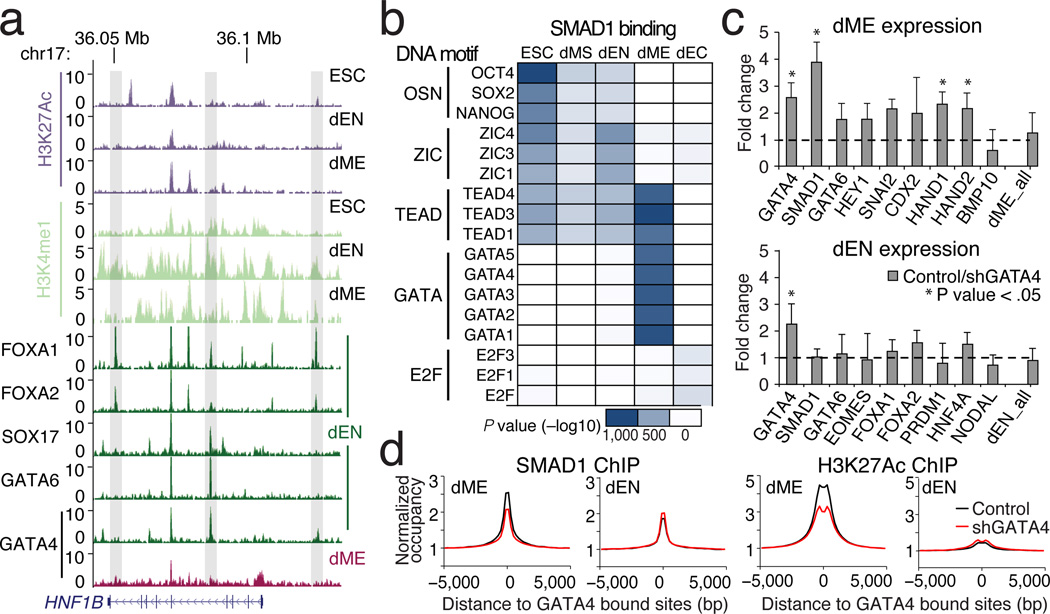
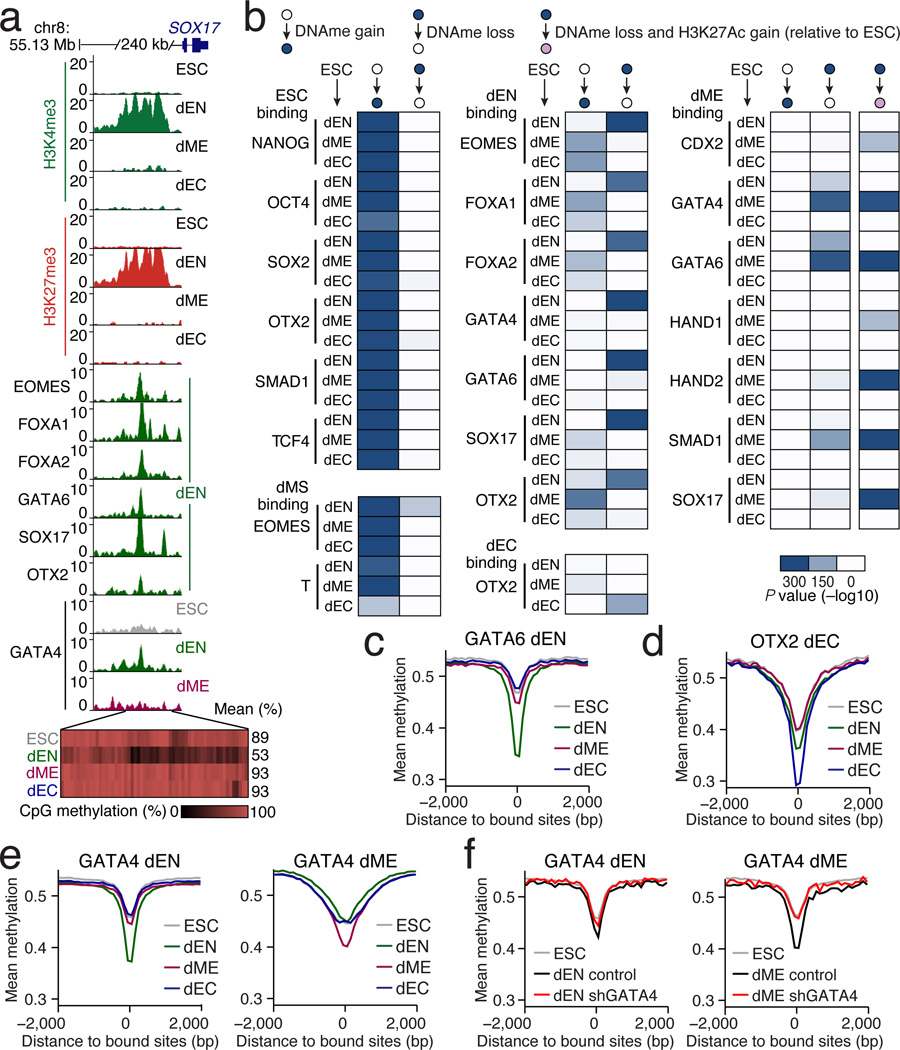
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
