

Current Understanding of the Role of Complement in IgA Nephropathy
- PMID: 25694468
- PMCID: PMC4483595
- DOI: 10.1681/ASN.2014101000
Current Understanding of the Role of Complement in IgA Nephropathy
Abstract
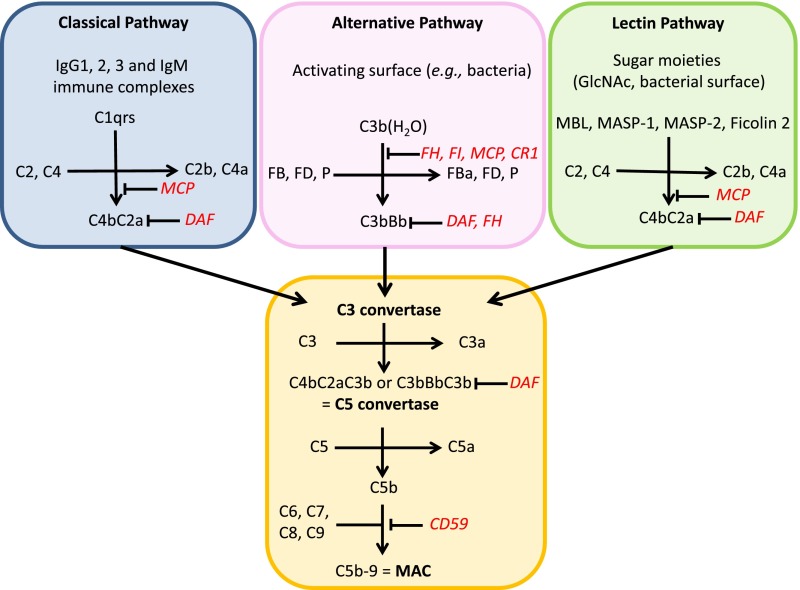
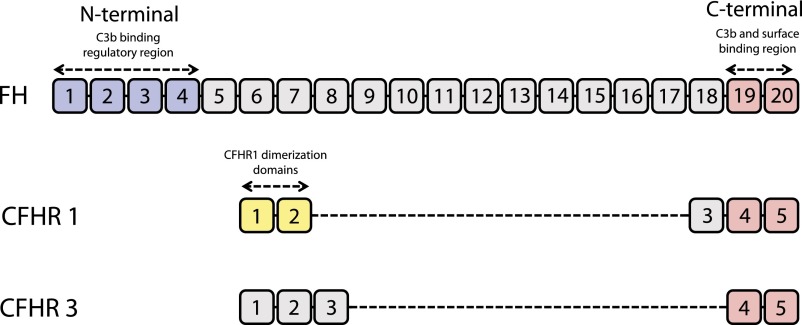
Complement activation has a role in the pathogenesis of IgA nephropathy, an autoimmune disease mediated by pathogenic immune complexes consisting of galactose-deficient IgA1 bound by antiglycan antibodies. Of three complement-activation pathways, the alternative and lectin pathways are involved in IgA nephropathy. IgA1 can activate both pathways in vitro, and pathway components are present in the mesangial immunodeposits, including properdin and factor H in the alternative pathway and mannan-binding lectin, mannan-binding lectin-associated serine proteases 1 and 2, and C4d in the lectin pathway. Genome-wide association studies identified deletion of complement factor H-related genes 1 and 3 as protective against the disease. Because the corresponding gene products compete with factor H in the regulation of the alternative pathway, it has been hypothesized that the absence of these genes could lead to more potent inhibition of complement by factor H. Complement activation can take place directly on IgA1-containing immune complexes in circulation and/or after their deposition in the mesangium. Notably, complement factors and their fragments may serve as biomarkers of IgA nephropathy in serum, urine, or renal tissue. A better understanding of the role of complement in IgA nephropathy may provide potential targets and rationale for development of complement-targeting therapy of the disease.
Keywords: IgA nephropathy; complement; immune complexes.
Copyright © 2015 by the American Society of Nephrology.
Figures



References
-
- Berger J, Hinglais N: Intercapillary deposits of IgA-IgG. J Urol Nephrol (Paris) 74: 694–695, 1968 - PubMed
-
- Berthoux FC, Mohey H, Afiani A: Natural history of primary IgA nephropathy. Semin Nephrol 28: 4–9, 2008 - PubMed
-
- Russell MW, Mestecky J, Julian BA, Galla JH: IgA-associated renal diseases: Antibodies to environmental antigens in sera and deposition of immunoglobulins and antigens in glomeruli. J Clin Immunol 6: 74–86, 1986 - PubMed
-
- Jennette JC: The immunohistology of IgA nephropathy. Am J Kidney Dis 12: 348–352, 1988 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
