

Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia
- PMID: 25694517
- PMCID: PMC4400760
- DOI: 10.1128/JCM.02898-14
Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia
Abstract
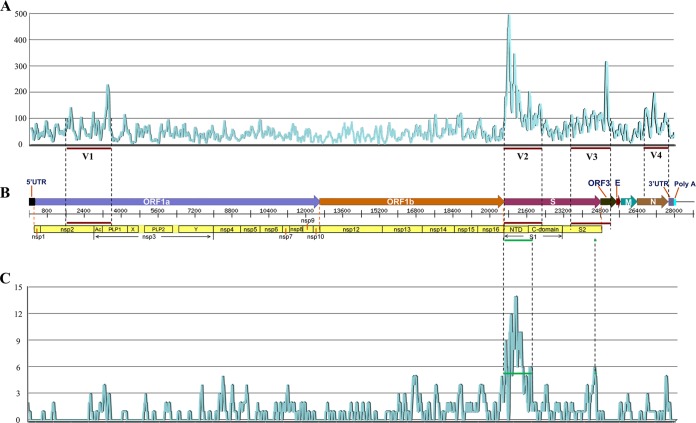
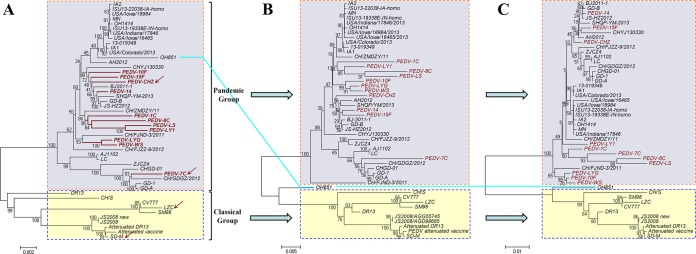
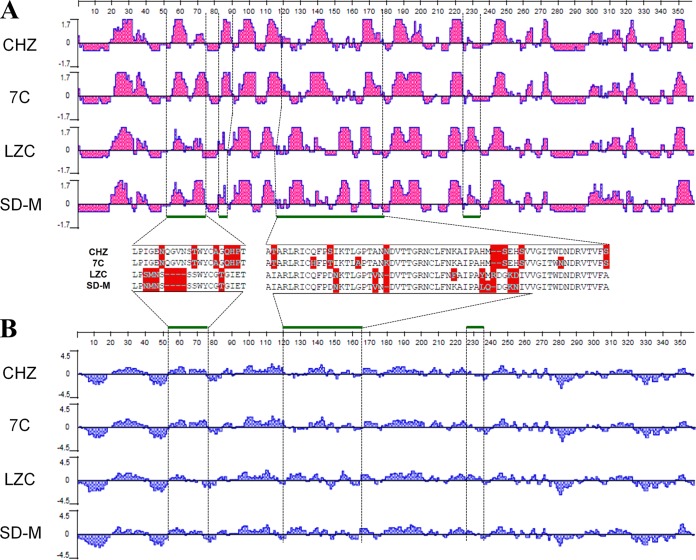
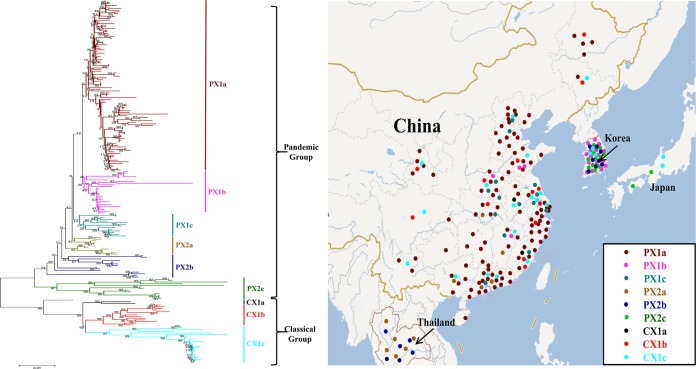
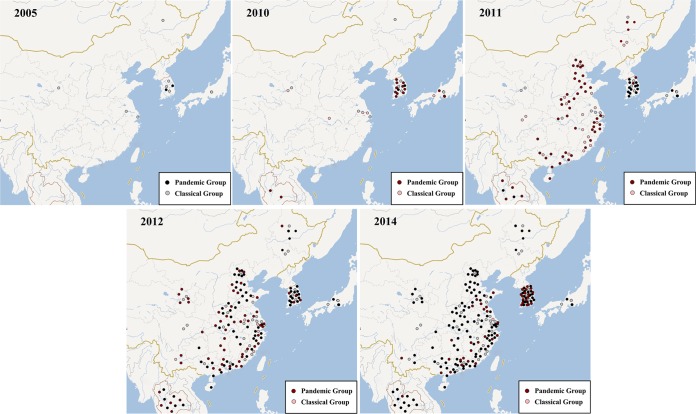
Porcine epidemic diarrhea has become pandemic in the Asian pig-breeding industry, causing significant economic loss. In the present study, 11 complete genomes of porcine epidemic diarrhea virus (PEDV) field isolates from China were determined and analyzed. Frequently occurring mutations were observed, which suggested that full understanding of the genomic and epidemiological characteristics is critical in the fight against PEDV epidemics. Comparative analysis of 49 available genomes clustered the PEDV strains into pandemic (PX) and classical (CX) groups and identified four hypervariable regions (V1 to V4). Further study indicated key roles for the spike (S) gene and the V2 region in distinguishing between the PX and CX groups and for studying genetic evolution. Genotyping and phylogeny-based geographical dissection based on 219 S genes revealed the complexity and severity of PEDV epidemics in Asia. Many subgroups have formed, with a wide array of mutations in different countries, leading to the outbreak of PEDV in Asia. Spatiotemporal reconstruction based on the analysis suggested that the pandemic group strains originated from South Korea and then extended into Japan, Thailand, and China. However, the novel pandemic strains in South Korea that appeared after 2013 may have originated from a Chinese variant. Thus, the serious PED epidemics in China and South Korea in recent years were caused by the complex subgroups of PEDV. The data in this study have important implications for understanding the ongoing PEDV outbreaks in Asia and will guide future efforts to effectively prevent and control PEDV.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Molecular characteristics of the spike gene of porcine epidemic diarrhoea virus strains in Eastern China in 2016.Virus Res. 2018 Mar 2;247:47-54. doi: 10.1016/j.virusres.2018.01.013. Epub 2018 Feb 3. Virus Res. 2018. PMID: 29412159
-
Genetic properties of endemic Chinese porcine epidemic diarrhea virus strains isolated since 2010.Arch Virol. 2013 Dec;158(12):2487-94. doi: 10.1007/s00705-013-1767-7. Arch Virol. 2013. PMID: 23797760 Free PMC article.
-
Detection and phylogenetic analyses of spike genes in porcine epidemic diarrhea virus strains circulating in China in 2016-2017.Virol J. 2017 Oct 10;14(1):194. doi: 10.1186/s12985-017-0860-z. Virol J. 2017. PMID: 29017599 Free PMC article.
-
Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus.Virol J. 2015 Dec 22;12:193. doi: 10.1186/s12985-015-0421-2. Virol J. 2015. PMID: 26689811 Free PMC article. Review.
-
Porcine epidemic diarrhea in China.Virus Res. 2016 Dec 2;226:7-13. doi: 10.1016/j.virusres.2016.05.026. Epub 2016 May 31. Virus Res. 2016. PMID: 27261169 Free PMC article. Review.
Cited by
-
Phylogenetic Analysis of Porcine Epidemic Diarrhea Virus (PEDV) during 2020-2022 and Isolation of a Variant Recombinant PEDV Strain.Int J Mol Sci. 2024 Oct 10;25(20):10878. doi: 10.3390/ijms252010878. Int J Mol Sci. 2024. PMID: 39456662 Free PMC article.
-
Porcine epidemic diarrhea virus strain CH/HLJ/18 isolated in China: characterization and phylogenetic analysis.Virol J. 2024 Jan 24;21(1):28. doi: 10.1186/s12985-023-02233-6. Virol J. 2024. PMID: 38268010 Free PMC article.
-
Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains.Virus Res. 2016 Dec 2;226:20-39. doi: 10.1016/j.virusres.2016.05.023. Epub 2016 Jun 8. Virus Res. 2016. PMID: 27288724 Free PMC article. Review.
-
Natural Evolution of Porcine Epidemic Diarrhea Viruses Isolated from Maternally Immunized Piglets.Animals (Basel). 2023 May 26;13(11):1766. doi: 10.3390/ani13111766. Animals (Basel). 2023. PMID: 37889642 Free PMC article.
-
Hypomethylated interferon regulatory factor 8 recruits activating protein-2α to attenuate porcine epidemic diarrhea virus infection in porcine jejunum.Front Immunol. 2023 Aug 1;14:1187144. doi: 10.3389/fimmu.2023.1187144. eCollection 2023. Front Immunol. 2023. PMID: 37593742 Free PMC article.
References
-
- Have P, Moving V, Svansson V, Uttenthal A, Bloch B. 1992. Coronavirus infection in mink (Mustela vison): serological evidence of infection with a coronavirus related to transmissible gastroenteritis virus and porcine epidemic diarrhea virus. Vet Microbiol 31:1–10. doi:10.1016/0378-1135(92)90135-G. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
