

Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection
- PMID: 25697341
- PMCID: PMC4408413
- DOI: 10.1093/molbev/msv022
Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection
Abstract
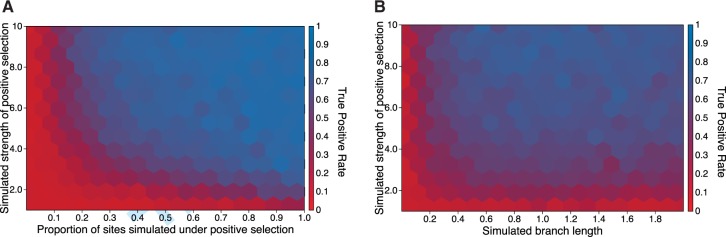
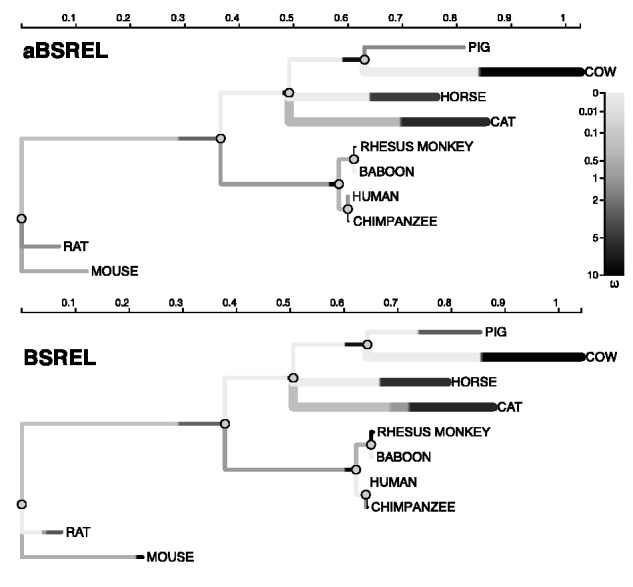
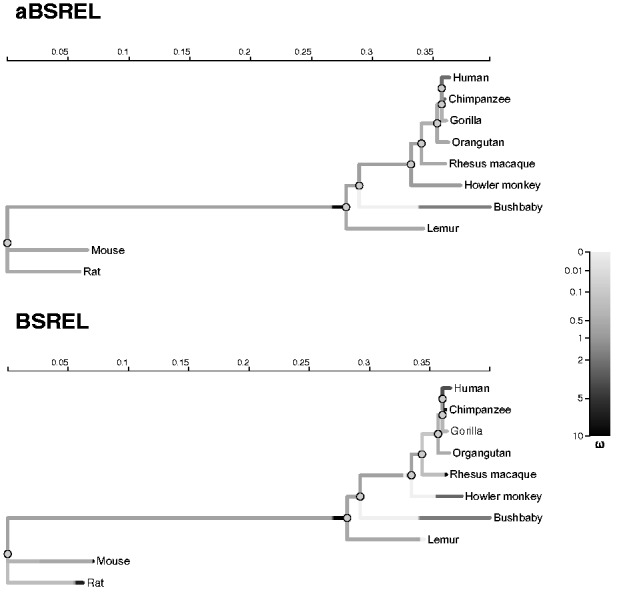
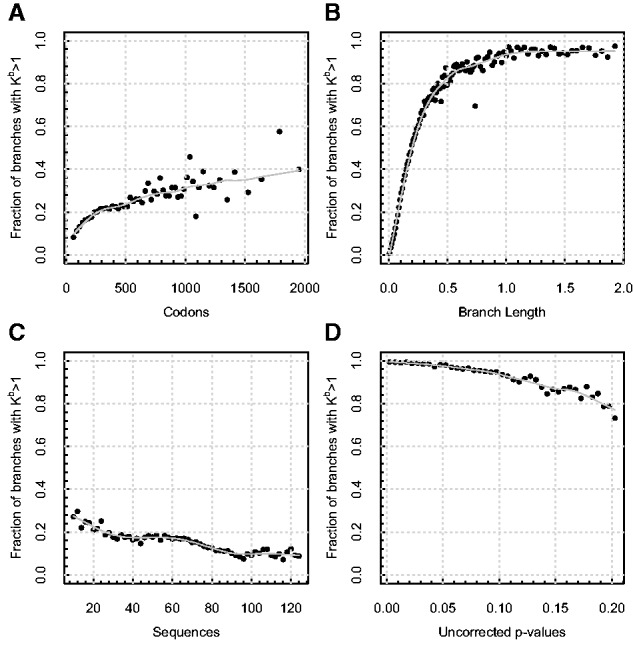
Over the past two decades, comparative sequence analysis using codon-substitution models has been honed into a powerful and popular approach for detecting signatures of natural selection from molecular data. A substantial body of work has focused on developing a class of "branch-site" models which permit selective pressures on sequences, quantified by the ω ratio, to vary among both codon sites and individual branches in the phylogeny. We develop and present a method in this class, adaptive branch-site random effects likelihood (aBSREL), whose key innovation is variable parametric complexity chosen with an information theoretic criterion. By applying models of different complexity to different branches in the phylogeny, aBSREL delivers statistical performance matching or exceeding best-in-class existing approaches, while running an order of magnitude faster. Based on simulated data analysis, we offer guidelines for what extent and strength of diversifying positive selection can be detected reliably and suggest that there is a natural limit on the optimal parametric complexity for "branch-site" models. An aBSREL analysis of 8,893 Euteleostomes gene alignments demonstrates that over 80% of branches in typical gene phylogenies can be adequately modeled with a single ω ratio model, that is, current models are unnecessarily complicated. However, there are a relatively small number of key branches, whose identities are derived from the data using a model selection procedure, for which it is essential to accurately model evolutionary complexity.
Keywords: branch-site model; episodic selection; evolutionary model; model complexity; random effects model; variable selection.
© The Author 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Aguileta G, Refrégier G, Yockteng R, Fournier E, Giraud T. Rapidly evolving genes in pathogens: methods for detecting positive selection and examples among fungi, bacteria, viruses and protists. Infect Genet Evol. 2009;9:656–670. - PubMed
-
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18:1585–1592. - PubMed
-
- Anisimova M, Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 2009;26:255–271. - PubMed
-
- Anisimova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007;24:1219–1228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K24 AI100665/AI/NIAID NIH HHS/United States
- U54 HL108460/HL/NHLBI NIH HHS/United States
- AI36214/AI/NIAID NIH HHS/United States
- U54HL108460/HL/NHLBI NIH HHS/United States
- GM093939/GM/NIGMS NIH HHS/United States
- R01 GM093939/GM/NIGMS NIH HHS/United States
- DP1 DA034978/DA/NIDA NIH HHS/United States
- DA034978/DA/NIDA NIH HHS/United States
- K01 AI110181/AI/NIAID NIH HHS/United States
- U01 GM110749/GM/NIGMS NIH HHS/United States
- AI090970/AI/NIAID NIH HHS/United States
- U19 AI090970/AI/NIAID NIH HHS/United States
- AI110181/AI/NIAID NIH HHS/United States
- U01GM110749/GM/NIGMS NIH HHS/United States
- P30 AI036214/AI/NIAID NIH HHS/United States
- AI100665/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
