

The multifaceted role of the vasculature in endochondral fracture repair
- PMID: 25699016
- PMCID: PMC4318416
- DOI: 10.3389/fendo.2015.00004
The multifaceted role of the vasculature in endochondral fracture repair
Abstract
Fracture healing is critically dependent upon an adequate vascular supply. The normal rate for fracture delayed or non-union is estimated to be between 10 and 15%, and annual fracture numbers are approximately 15 million cases per year. However, when there is decreased vascular perfusion to the fracture, incidence of impaired healing rises dramatically to 46%. Reduction in the blood supply to the fracture can be the result of traumatic injuries that physically disrupt the vasculature and damage supportive soft tissue, the result of anatomical location (i.e., distal tibia), or attributed to physiological conditions such as age, diabetes, or smoking. The role of the vasculature during repair is multifaceted and changes during the course of healing. In this article, we review recent insights into the role of the vasculature during fracture repair. Taken together these data highlight the need for an updated model for endochondral repair to facilitate improved therapeutic approaches to promote bone healing.
Keywords: angiogenesis; bone biology; cartilage transformation; endochondral ossification; fracture repair.
Figures
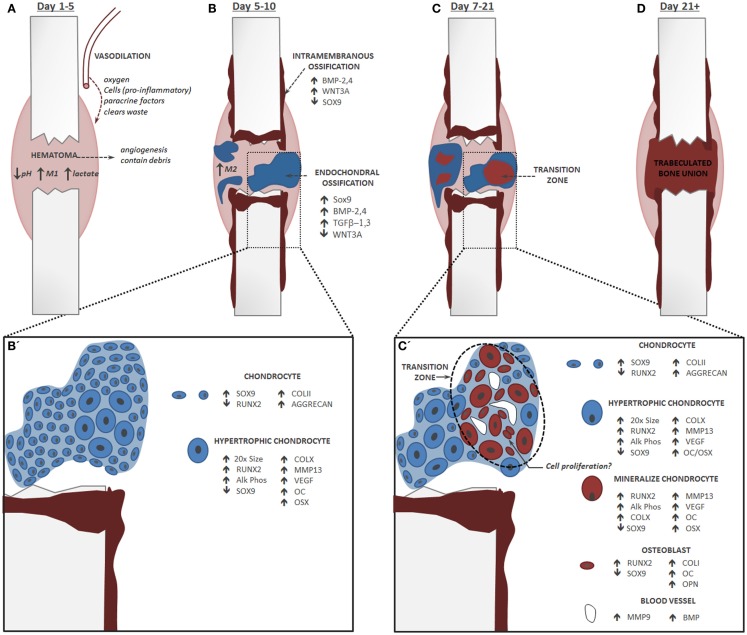
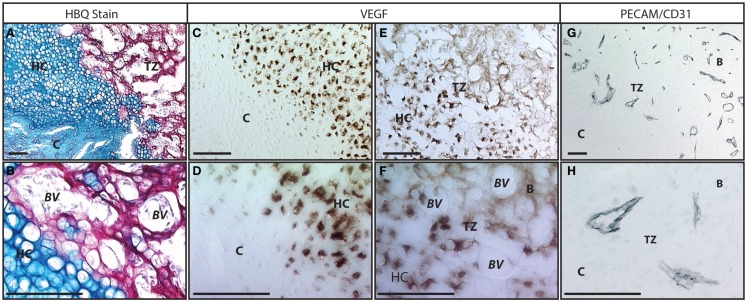
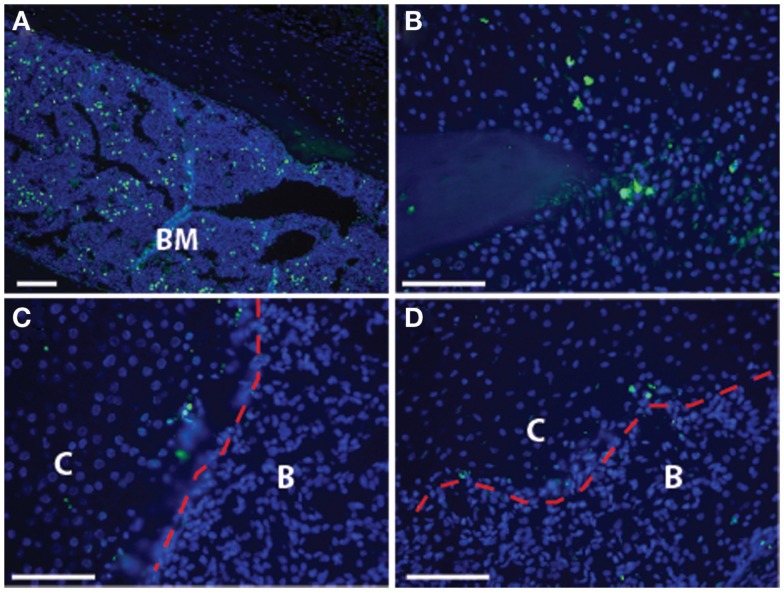
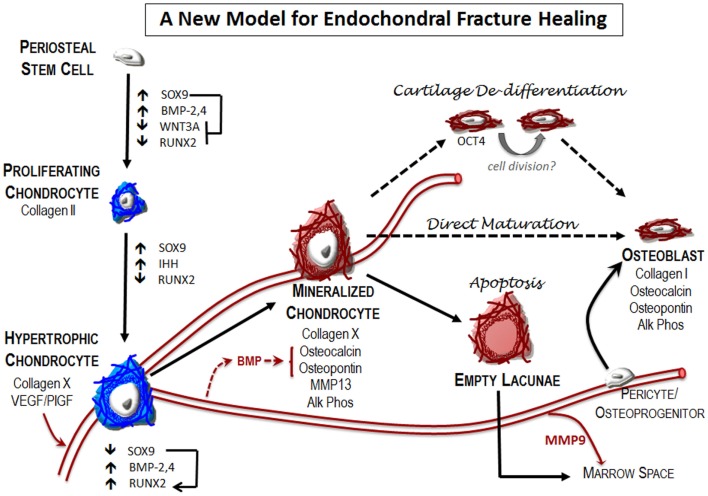
References
-
- Dickson KF, Katzman S, Paiement G. The importance of the blood supply in the healing of tibial fractures. Contemp Orthop (1995) 30:489–93. - PubMed
-
- Available Hospital Based Emergency Care: At the Breaking Point. Washington, DC: The National Acadamies Press; (2007).
-
- The Burden of Musculoskeletal Diseases in the United States. Rosemont, IL: American Academy of Orthopaedic Surgeons; (2008).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources