

Disease networks. Uncovering disease-disease relationships through the incomplete interactome
- PMID: 25700523
- PMCID: PMC4435741
- DOI: 10.1126/science.1257601
Disease networks. Uncovering disease-disease relationships through the incomplete interactome
Abstract
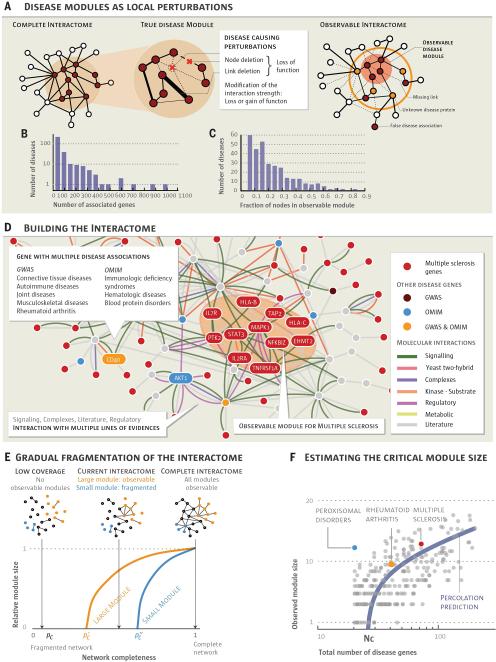
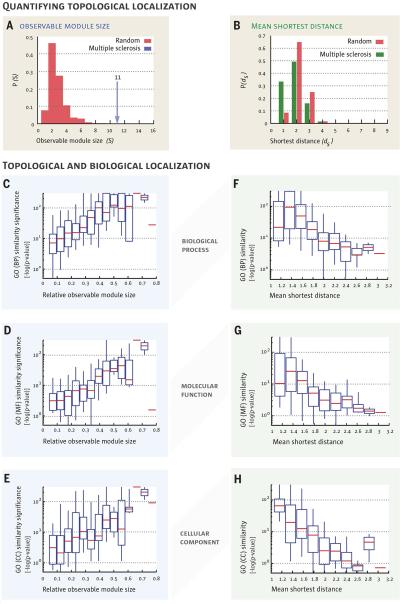
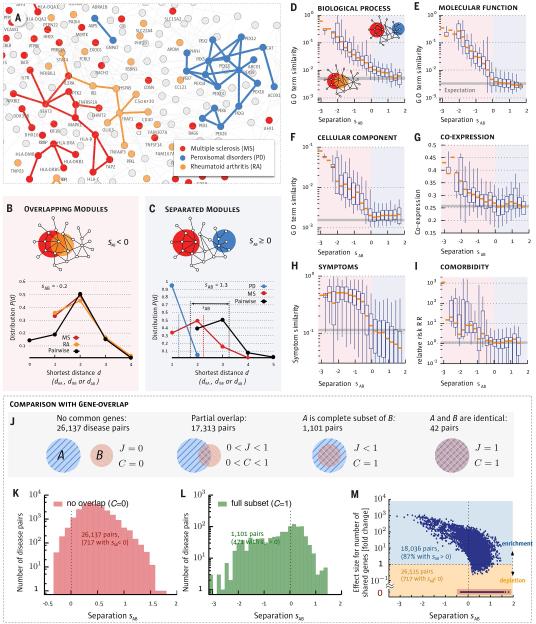
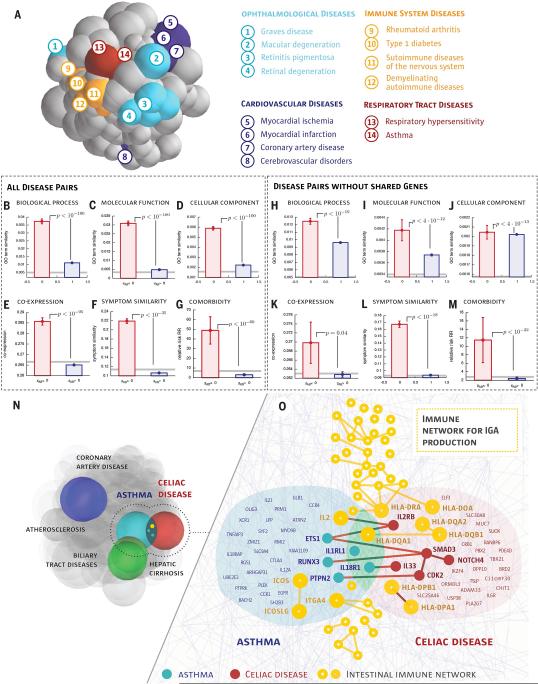
According to the disease module hypothesis, the cellular components associated with a disease segregate in the same neighborhood of the human interactome, the map of biologically relevant molecular interactions. Yet, given the incompleteness of the interactome and the limited knowledge of disease-associated genes, it is not obvious if the available data have sufficient coverage to map out modules associated with each disease. Here we derive mathematical conditions for the identifiability of disease modules and show that the network-based location of each disease module determines its pathobiological relationship to other diseases. For example, diseases with overlapping network modules show significant coexpression patterns, symptom similarity, and comorbidity, whereas diseases residing in separated network neighborhoods are phenotypically distinct. These tools represent an interactome-based platform to predict molecular commonalities between phenotypically related diseases, even if they do not share primary disease genes.
Copyright © 2015, American Association for the Advancement of Science.
Figures
References
Publication types
MeSH terms
Grants and funding
- U01 HL108630/HL/NHLBI NIH HHS/United States
- U01 HG007690/HG/NHGRI NIH HHS/United States
- P01 HL083069/HL/NHLBI NIH HHS/United States
- P01-HL083069/HL/NHLBI NIH HHS/United States
- R37-HL061795/HL/NHLBI NIH HHS/United States
- U01 HG001715/HG/NHGRI NIH HHS/United States
- U01-HG007690/HG/NHGRI NIH HHS/United States
- P50 HG004233/HG/NHGRI NIH HHS/United States
- R37 HL061795/HL/NHLBI NIH HHS/United States
- U01-HG001715/HG/NHGRI NIH HHS/United States
- U41 HG001715/HG/NHGRI NIH HHS/United States
- P50-HG004233/HG/NHGRI NIH HHS/United States
- U01-HL108630/HL/NHLBI NIH HHS/United States
- RC2-HL101543/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
