

Nothing to sneeze at: a dynamic and integrative computational model of an influenza A virion
- PMID: 25703376
- PMCID: PMC4353694
- DOI: 10.1016/j.str.2014.12.019
Nothing to sneeze at: a dynamic and integrative computational model of an influenza A virion
Abstract
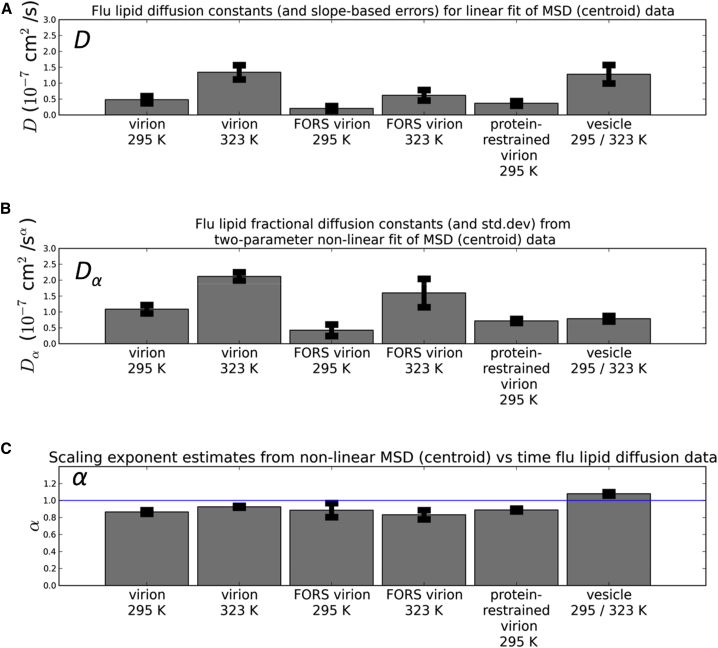
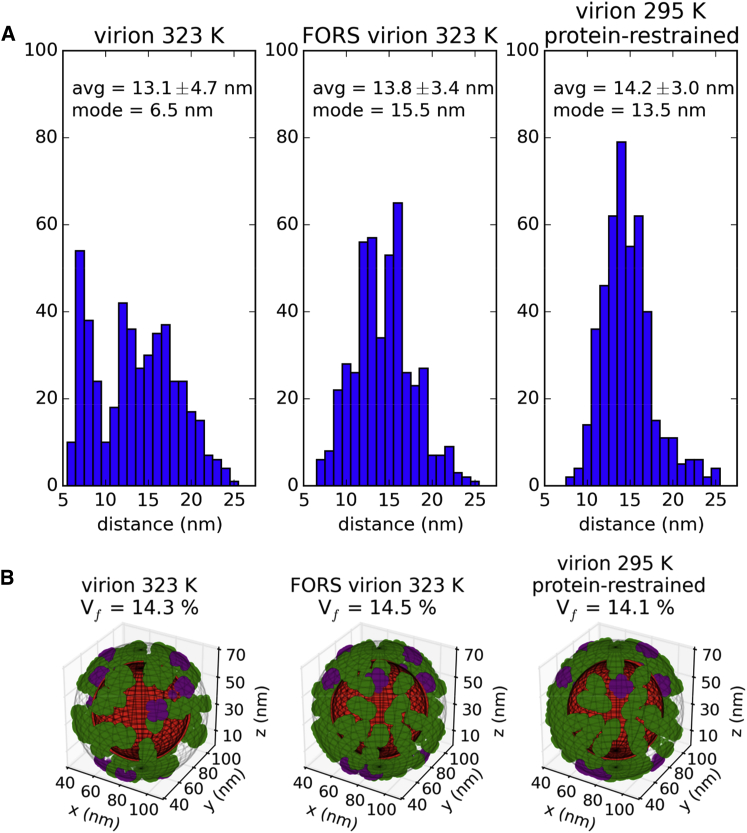
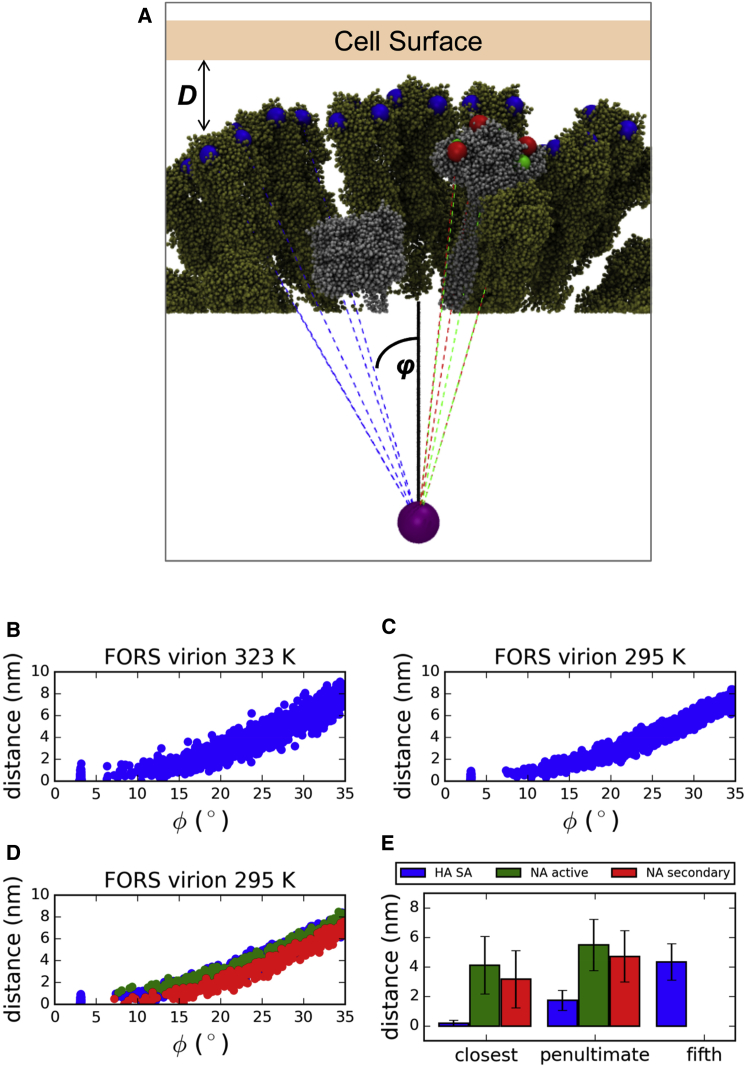
The influenza virus is surrounded by an envelope composed of a lipid bilayer and integral membrane proteins. Understanding the structural dynamics of the membrane envelope provides biophysical insights into aspects of viral function, such as the wide-ranging survival times of the virion in different environments. We have combined experimental data from X-ray crystallography, nuclear magnetic resonance spectroscopy, cryo-electron microscopy, and lipidomics to build a model of the intact influenza A virion. This is the basis of microsecond-scale coarse-grained molecular dynamics simulations of the virion, providing simulations at different temperatures and with varying lipid compositions. The presence of the Forssman glycolipid alters a number of biophysical properties of the virion, resulting in reduced mobility of bilayer lipid and protein species. Reduced mobility in the virion membrane may confer physical robustness to changes in environmental conditions. Our simulations indicate that viral spike proteins do not aggregate and thus are competent for multivalent immunoglobulin G interactions.
Copyright © 2015 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
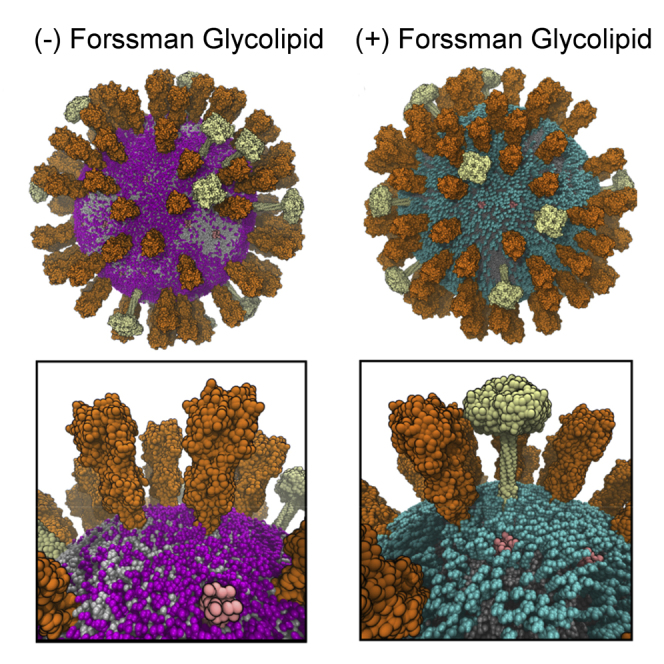
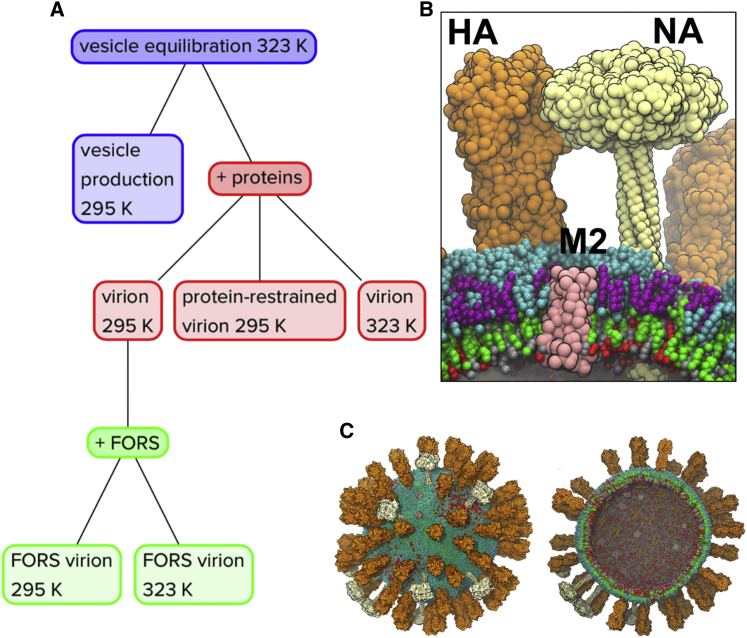
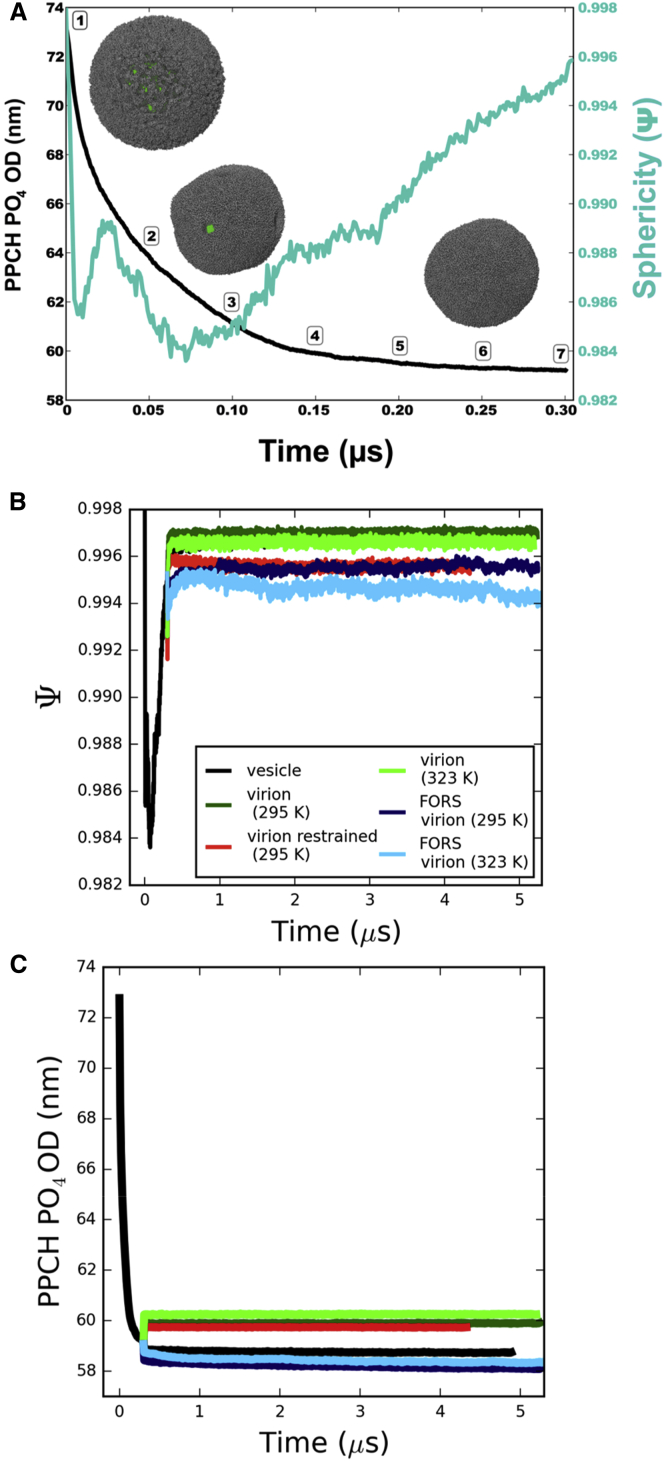
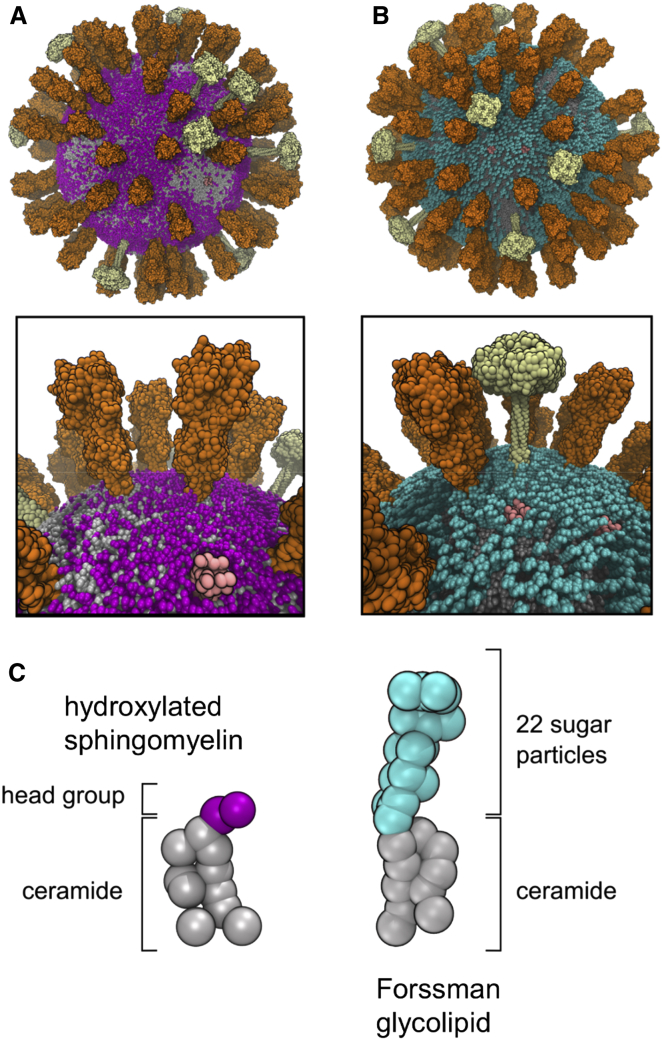
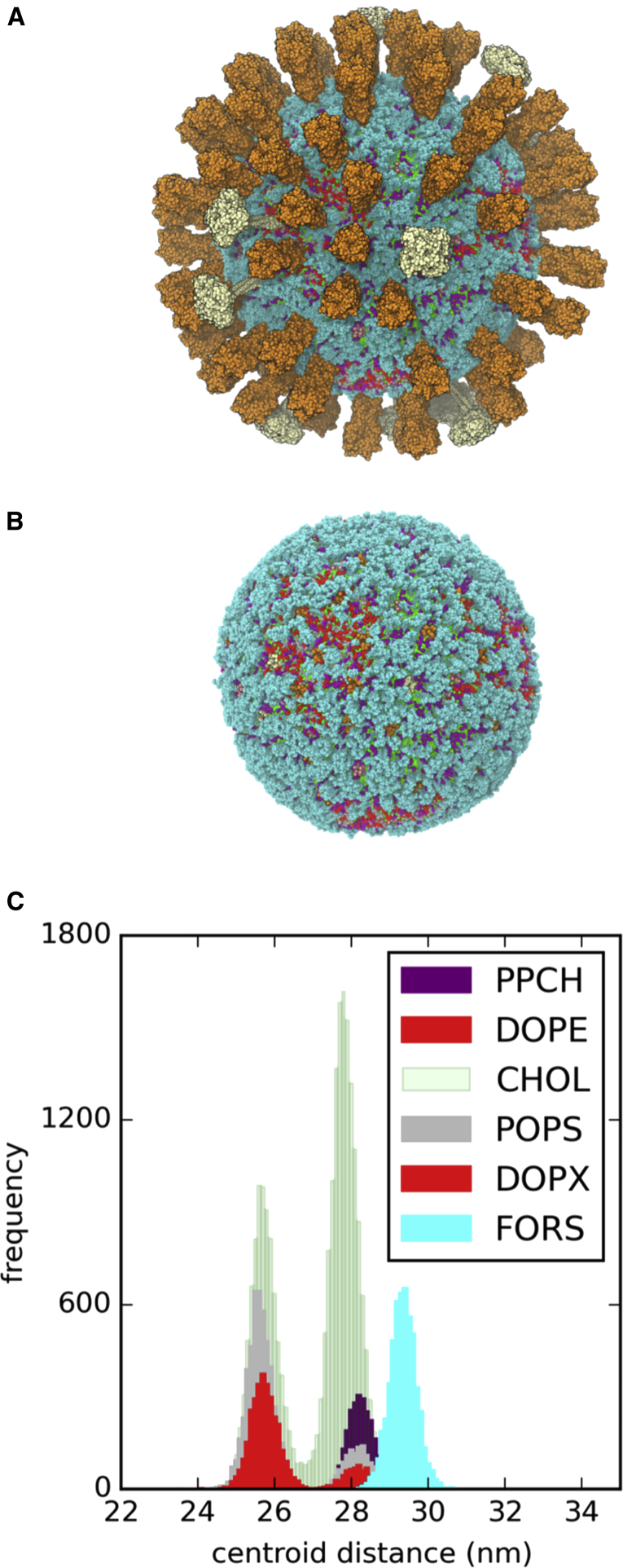
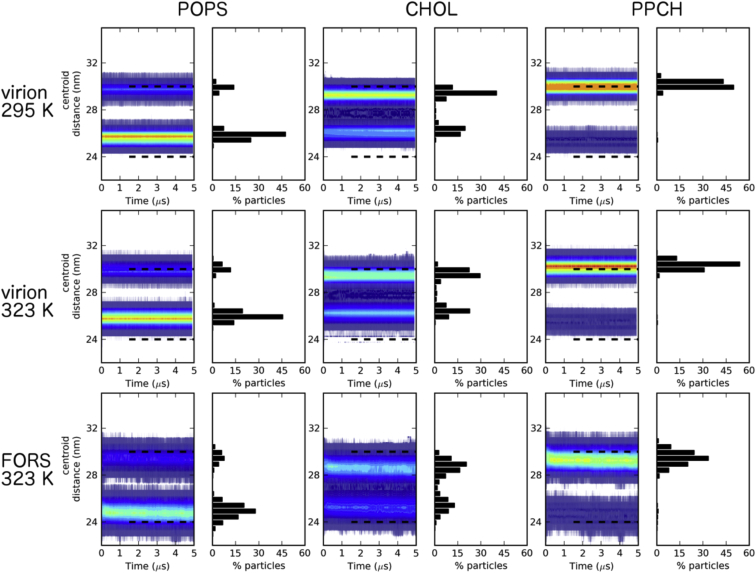
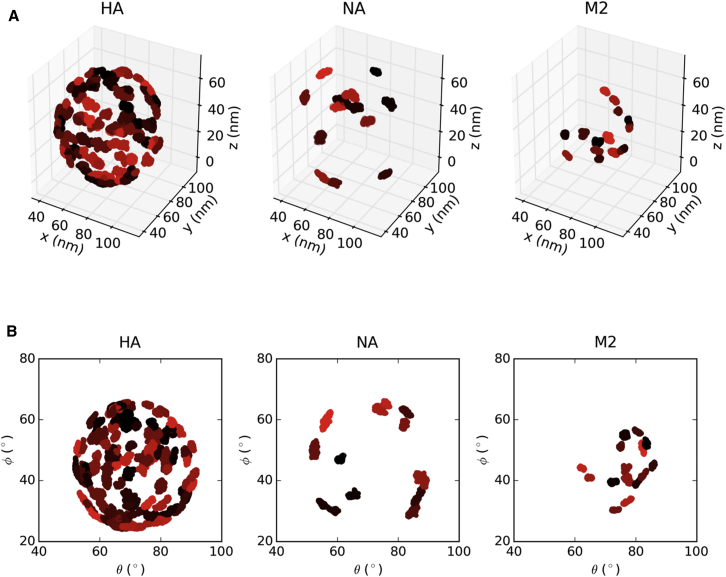
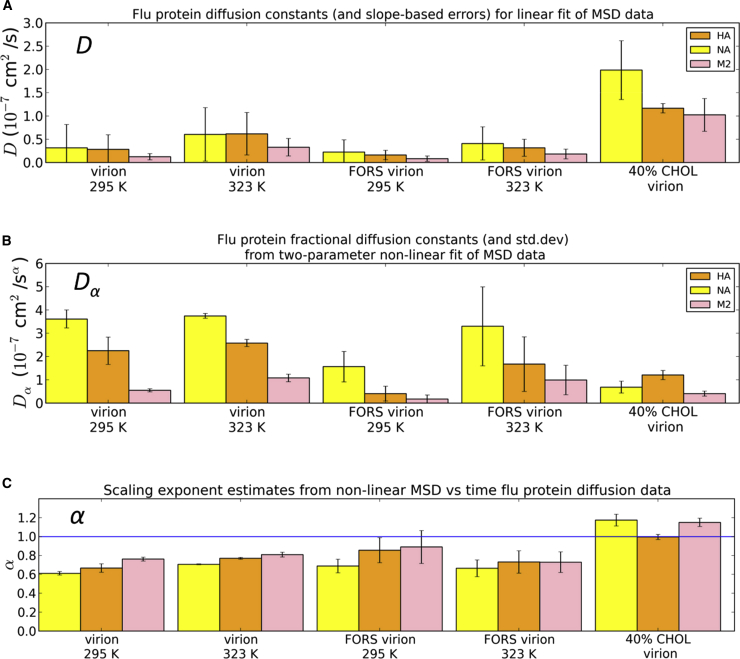
Comment in
-
Simulations move toward a cure for viral diseases.Structure. 2015 Mar 3;23(3):439-440. doi: 10.1016/j.str.2015.02.002. Structure. 2015. PMID: 25738384
Similar articles
-
The Role of the Membrane in the Structure and Biophysical Robustness of the Dengue Virion Envelope.Structure. 2016 Mar 1;24(3):375-82. doi: 10.1016/j.str.2015.12.011. Epub 2016 Jan 28. Structure. 2016. PMID: 26833387 Free PMC article.
-
Cold co-extraction of hemagglutinin and matrix M1 protein from influenza virus A by a combination of non-ionic detergents allows for visualization of the raft-like nature of the virus envelope.Arch Virol. 2008;153(10):1977-80. doi: 10.1007/s00705-008-0214-7. Epub 2008 Sep 30. Arch Virol. 2008. PMID: 18825482
-
Phosphatidylserine Lateral Organization Influences the Interaction of Influenza Virus Matrix Protein 1 with Lipid Membranes.J Virol. 2017 May 26;91(12):e00267-17. doi: 10.1128/JVI.00267-17. Print 2017 Jun 15. J Virol. 2017. PMID: 28356535 Free PMC article.
-
Supramolecular Architecture of the Coronavirus Particle.Adv Virus Res. 2016;96:1-27. doi: 10.1016/bs.aivir.2016.08.005. Epub 2016 Sep 15. Adv Virus Res. 2016. PMID: 27712621 Free PMC article. Review.
-
[Electron microscopic analysis of viral assembly and budding].Uirusu. 2009 Jun;59(1):99-106. doi: 10.2222/jsv.59.99. Uirusu. 2009. PMID: 19927994 Review. Japanese.
Cited by
-
Understanding Virus Structure and Dynamics through Molecular Simulations.J Chem Theory Comput. 2023 Jun 13;19(11):3025-3036. doi: 10.1021/acs.jctc.3c00116. Epub 2023 May 16. J Chem Theory Comput. 2023. PMID: 37192279 Free PMC article. Review.
-
Protein-driven membrane remodeling: Molecular perspectives from Flaviviridae infections.Biophys J. 2023 Jun 6;122(11):1890-1899. doi: 10.1016/j.bpj.2022.11.015. Epub 2022 Nov 11. Biophys J. 2023. PMID: 36369756 Free PMC article. Review.
-
Mesoscale All-Atom Influenza Virus Simulations Suggest New Substrate Binding Mechanism.ACS Cent Sci. 2020 Feb 26;6(2):189-196. doi: 10.1021/acscentsci.9b01071. Epub 2020 Feb 19. ACS Cent Sci. 2020. PMID: 32123736 Free PMC article.
-
Enabling Atomistic Modeling and Simulation of Complex Curved Cellular Membranes with xMAS Builder.bioRxiv [Preprint]. 2025 Jan 19:2025.01.14.632907. doi: 10.1101/2025.01.14.632907. bioRxiv. 2025. PMID: 39868109 Free PMC article. Preprint.
-
Breathing and tilting: mesoscale simulations illuminate influenza glycoprotein vulnerabilities.bioRxiv [Preprint]. 2022 Aug 7:2022.08.02.502576. doi: 10.1101/2022.08.02.502576. bioRxiv. 2022. Update in: ACS Cent Sci. 2022 Dec 28;8(12):1646-1663. doi: 10.1021/acscentsci.2c00981. PMID: 35982676 Free PMC article. Updated. Preprint.
References
-
- Almeida P.F.F., Vaz W.L.C. Lateral diffusion in membranes. In: Lipowsky R., Sackmann E., editors. Structure and Dynamics of Membranes: From Cells to Vesicles. Vol. 1. North-Holland; 1995. pp. 305–357. (Handbook of Biological Physics).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
