

The membrane- and soluble-protein helix-helix interactome: similar geometry via different interactions
- PMID: 25703378
- PMCID: PMC4351763
- DOI: 10.1016/j.str.2015.01.009
The membrane- and soluble-protein helix-helix interactome: similar geometry via different interactions
Abstract
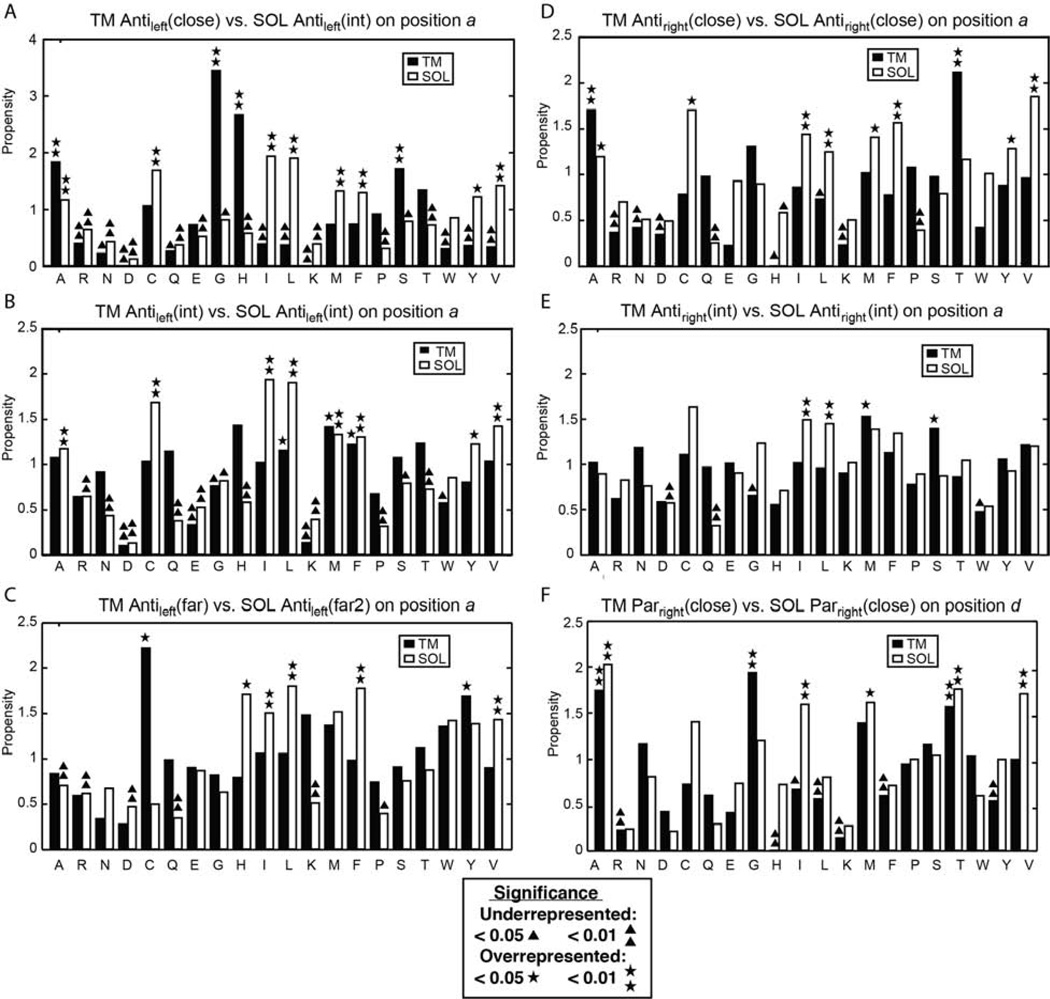
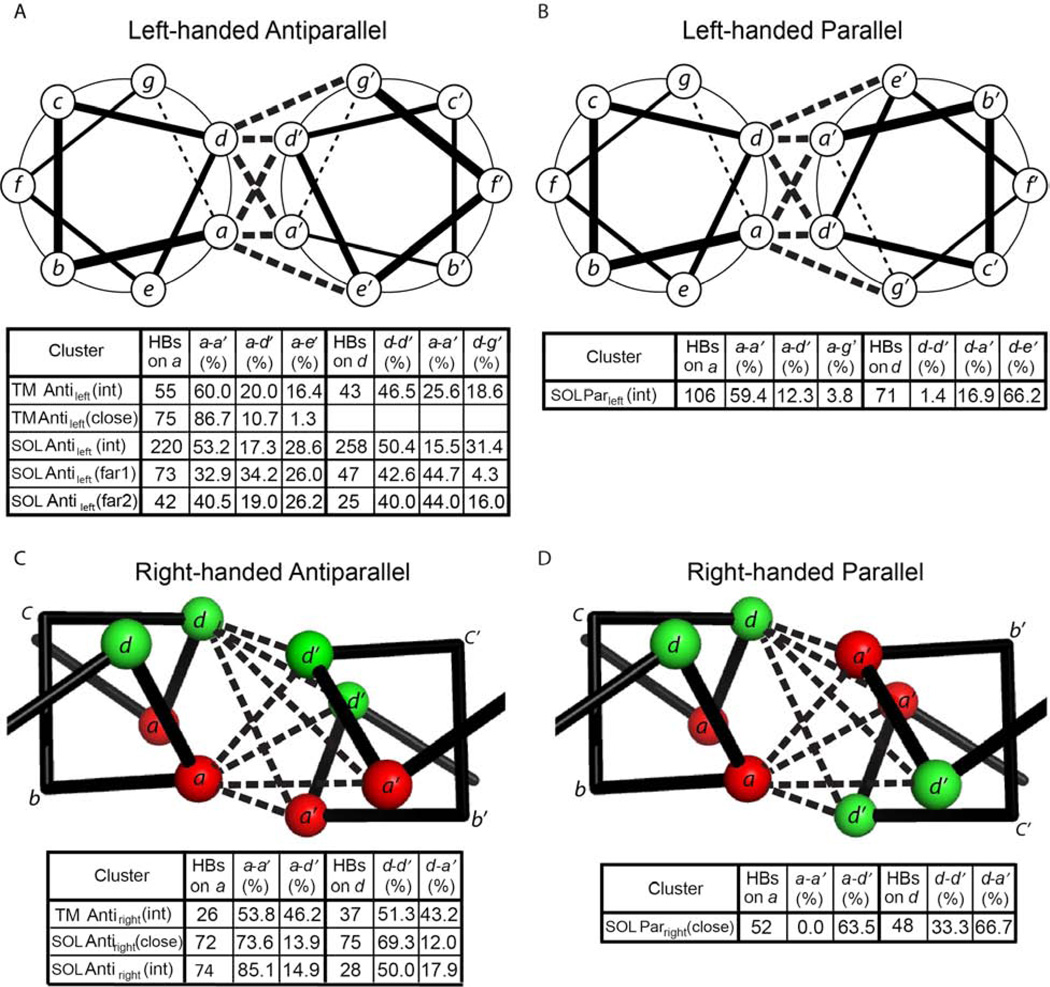
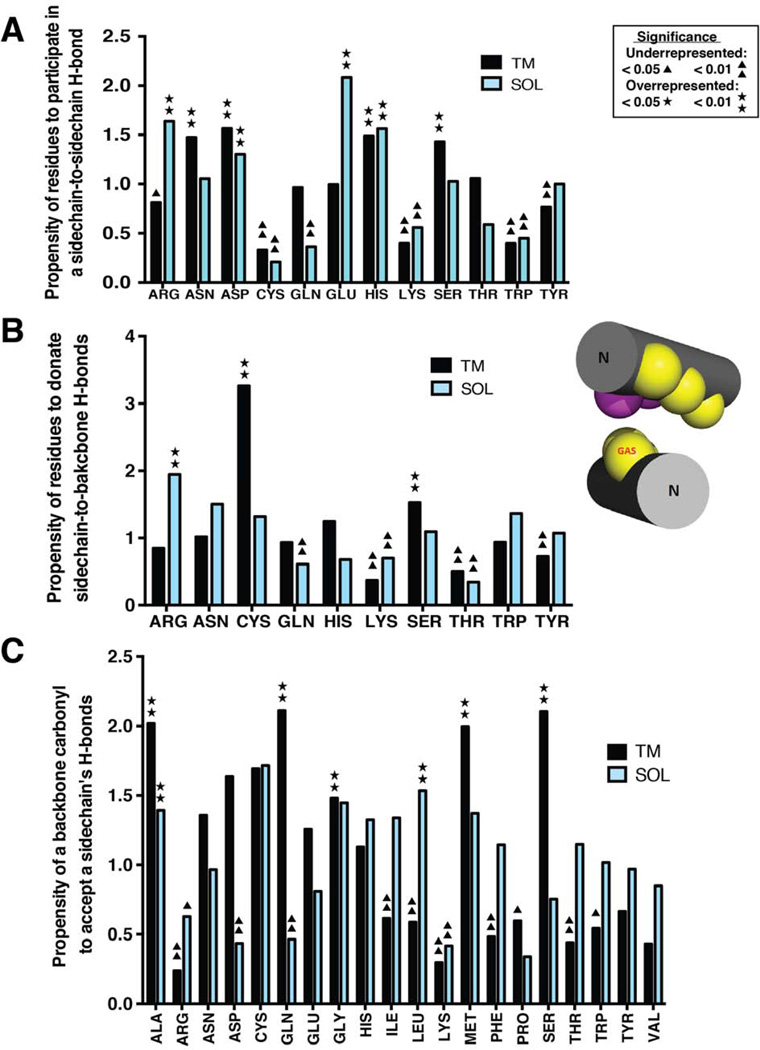
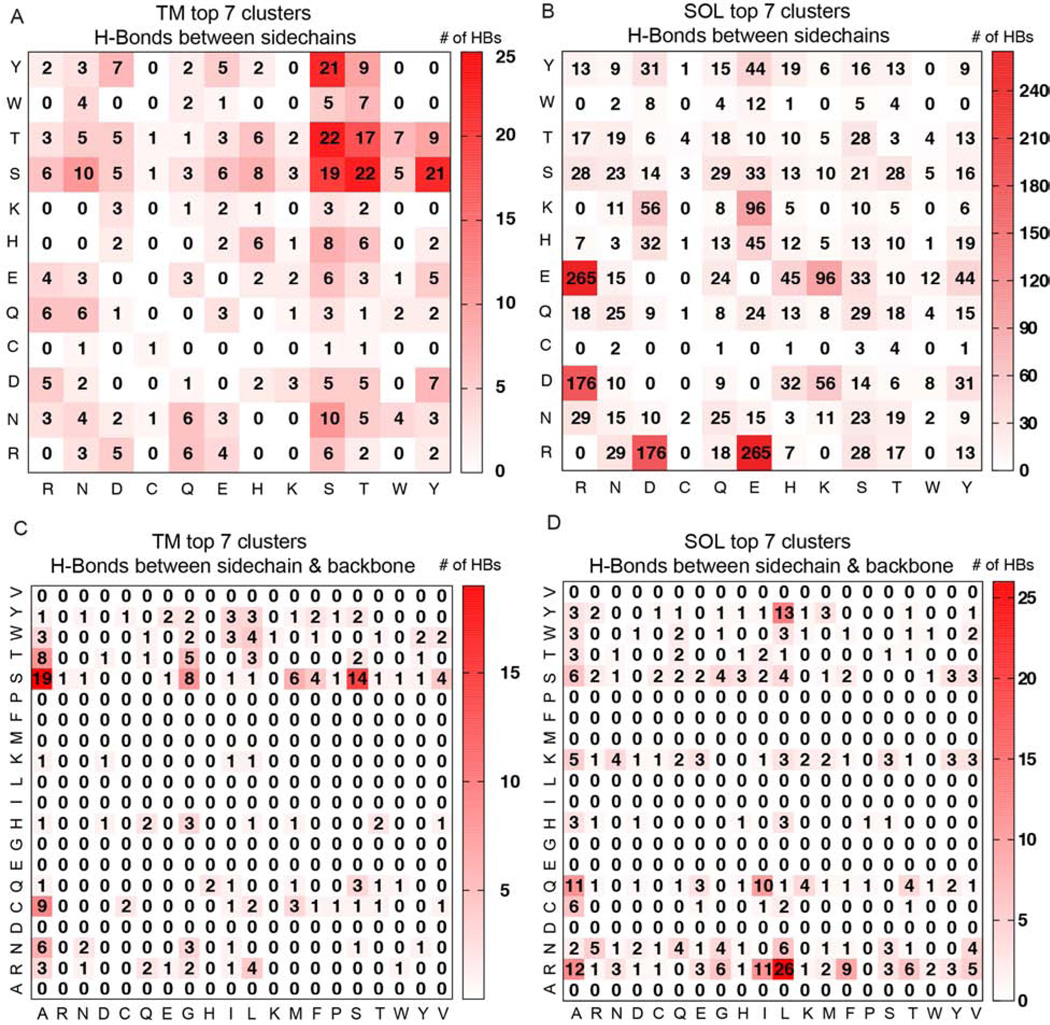
α Helices are a basic unit of protein secondary structure and therefore the interaction between helices is crucial to understanding tertiary and higher-order folds. Comparing subtle variations in the structural and sequence motifs between membrane and soluble proteins sheds light on the different constraints faced by each environment and elucidates the complex puzzle of membrane protein folding. Here, we demonstrate that membrane and water-soluble helix pairs share a small number of similar folds with various interhelical distances. The composition of the residues that pack at the interface between corresponding motifs shows that hydrophobic residues tend to be more enriched in the water-soluble class of structures and small residues in the transmembrane class. The latter group facilitates packing via sidechain- and backbone-mediated hydrogen bonds within the low-dielectric membrane milieu. The helix-helix interactome space, with its associated sequence preferences and accompanying hydrogen-bonding patterns, should be useful for engineering, prediction, and design of protein structure.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
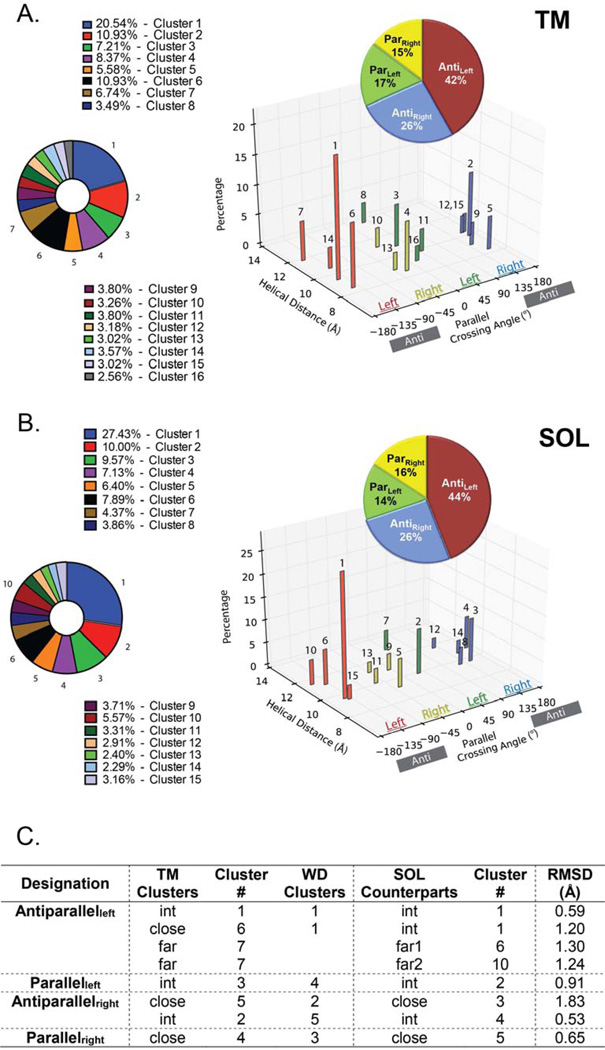
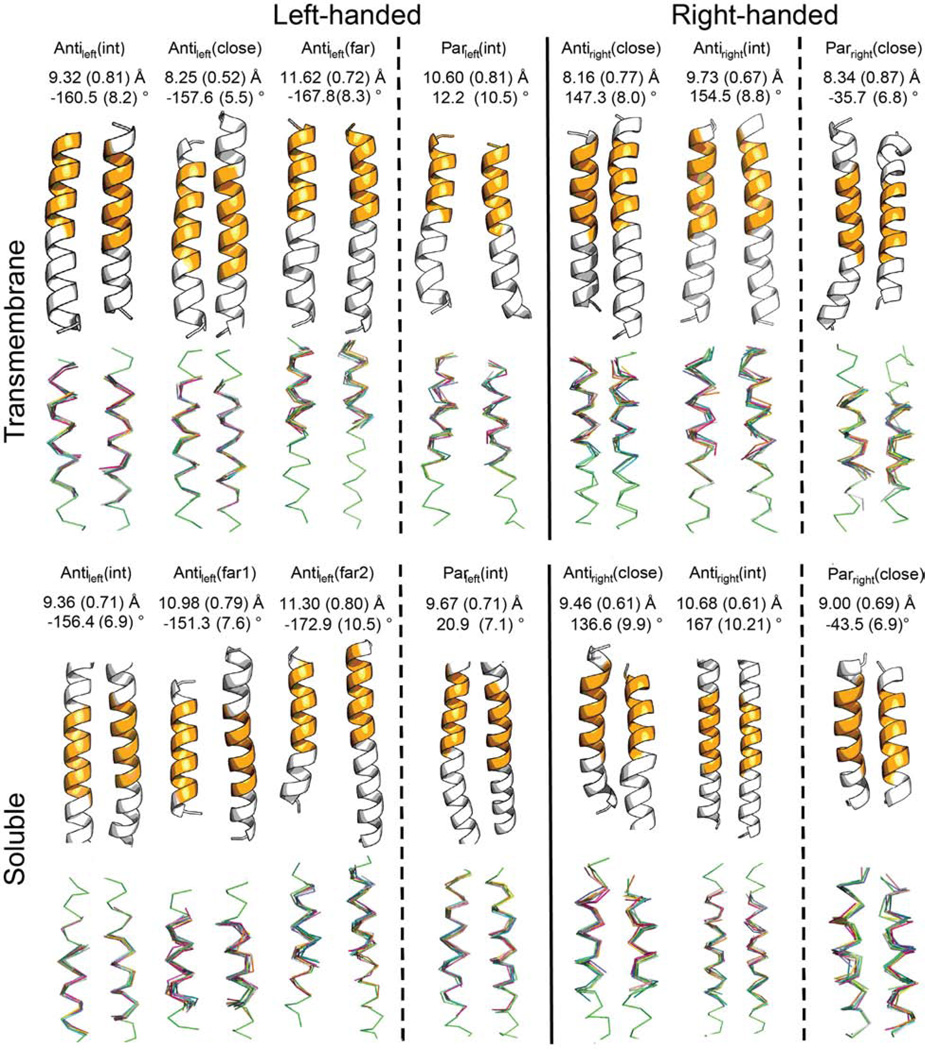
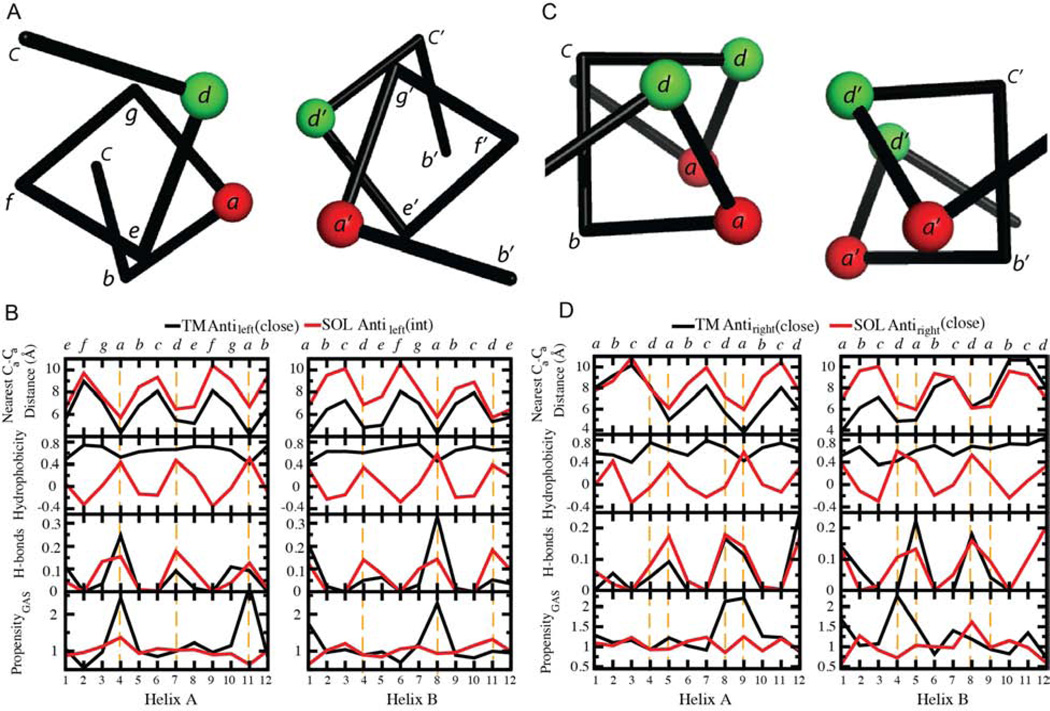
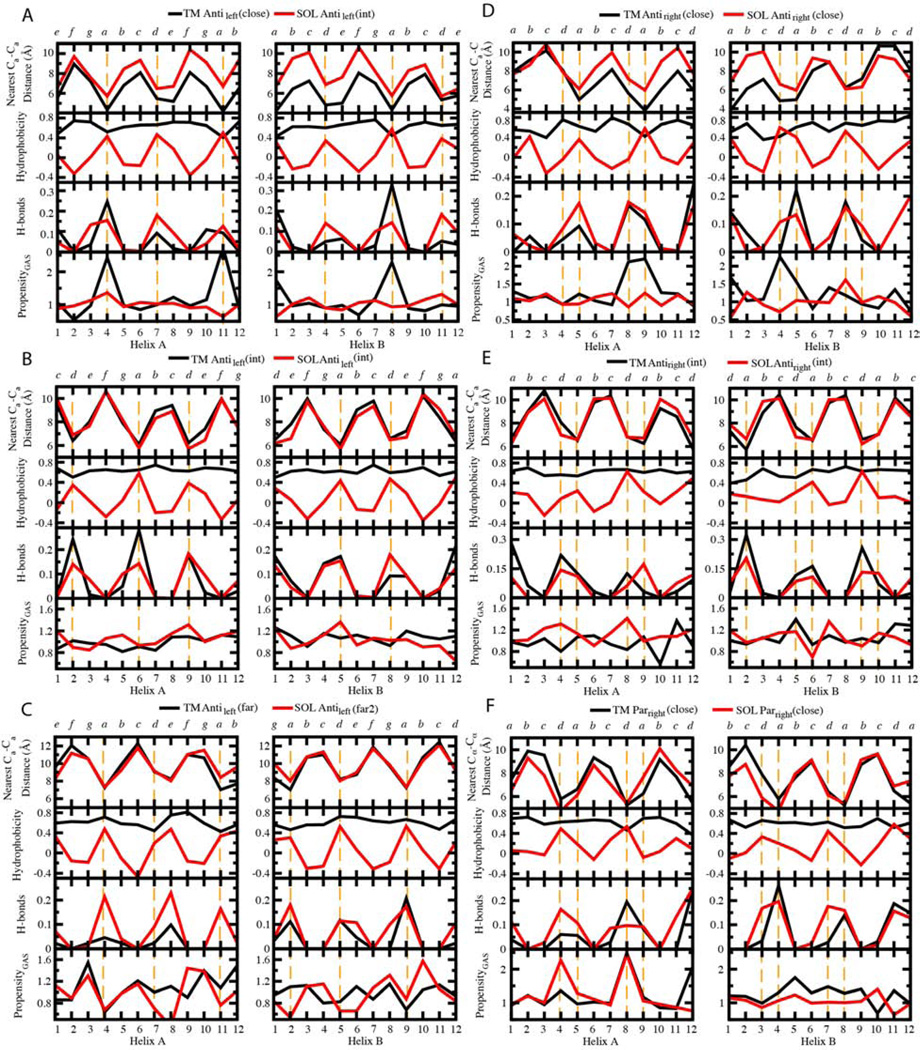
Comment in
-
Helix-helix interactions: is the medium the message?Structure. 2015 Mar 3;23(3):437-438. doi: 10.1016/j.str.2015.02.004. Structure. 2015. PMID: 25738383
References
-
- Acharya A, Rishi V, Vinson C. Stability of 100 homo and heterotypic coiled-coil a-a' pairs for ten amino acids (A, L, I, V, N, K, S, T, E, and R) Biochemistry. 2006;45:11324–11332. - PubMed
-
- Adamian L, Liang J. Interhelical hydrogen bonds and spatial motifs in membrane proteins: polar clamps and serine zippers. Proteins. 2002;47:209–218. - PubMed
-
- Baker E, Hubbard R. Hydrogen bonding in globular proteins. Progress in biophysics and molecular biology. 1984;44:97–179. - PubMed
-
- Bansal M, Kumar S, Velavan R. HELANAL: a program to characterize helix geometry in proteins. J Biomol Struct Dyn. 2000;17:811–819. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
