

New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics
- PMID: 25703571
- PMCID: PMC4571192
- DOI: 10.1111/imr.12255
New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics
Abstract
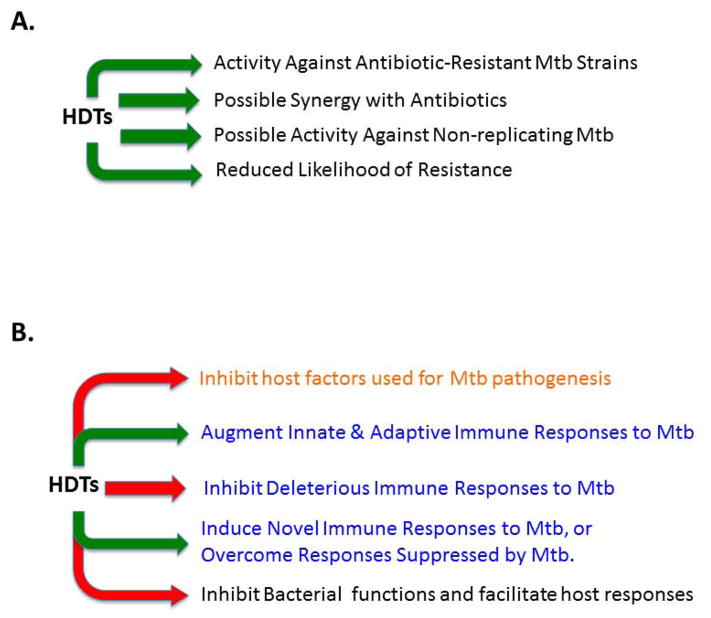
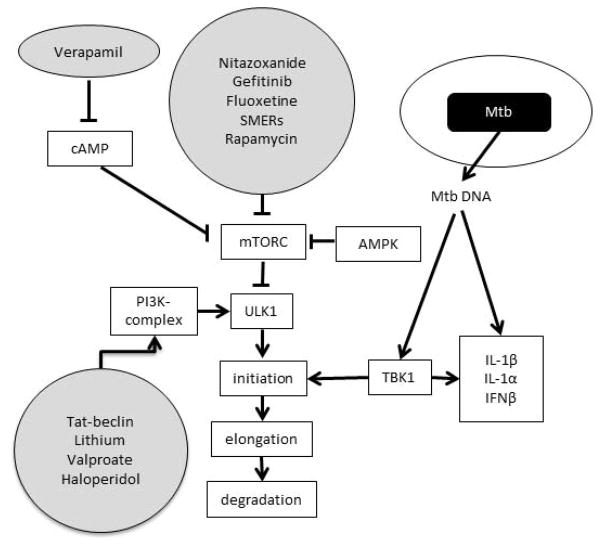
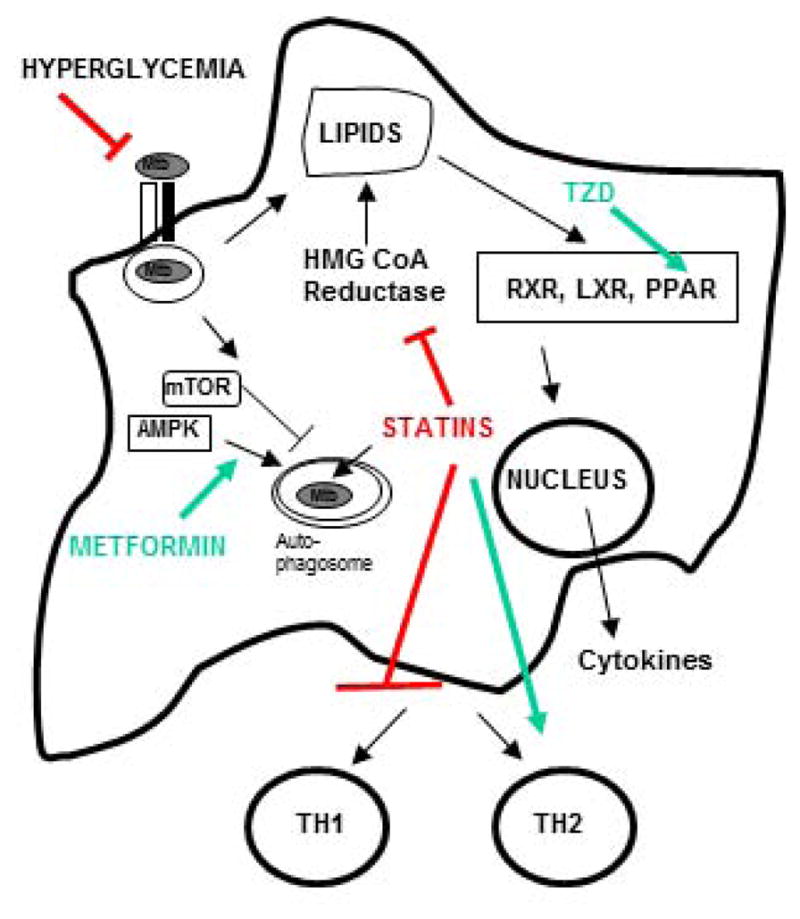
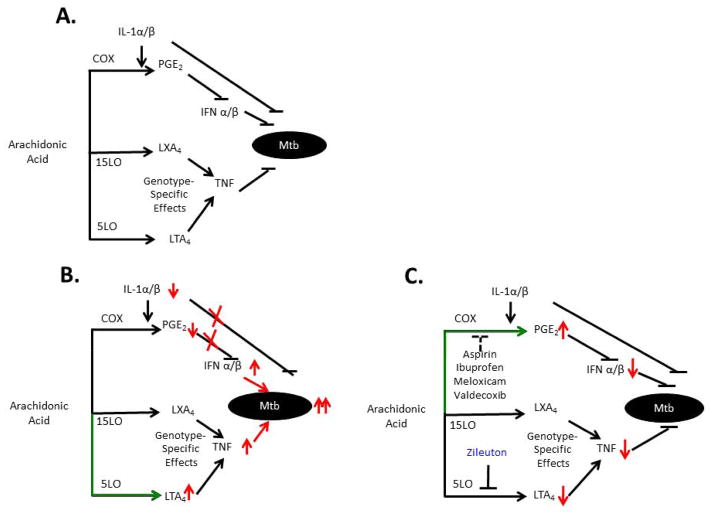
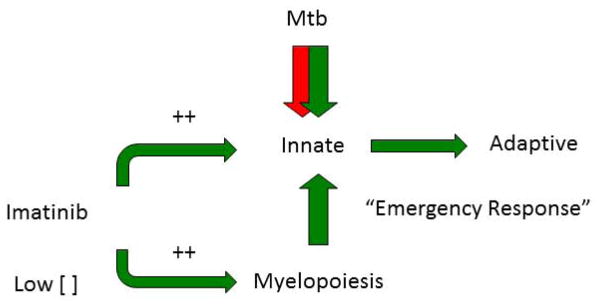
Despite the availability of Mycobacterium tuberculosis (Mtb) drugs for over 50 years, tuberculosis (TB) remains at pandemic levels. New drugs are urgently needed for resistant strains, shortening duration of treatment, and targeting different stages of the disease, especially for treatment during human immunodeficiency virus co-infection. One solution to the conundrum that antibiotics kill the bacillus yet select for resistance is to target the host rather than the pathogen. Here, we discuss recent progress in so-called 'host-directed therapeutics' (HDTs), focusing on two general mechanistic strategies: (i) HDTs that disrupt Mtb pathogenesis in macrophages and (ii) immunomodulatory HDTs that facilitate protective immune responses that kill Mtb or reduce deleterious responses that exacerbate disease. HDTs hold significant promise as adjunctive therapies in that they are less likely to engender resistance, will likely have efficacy against antibiotic-resistant strains, and may have activity against non-replicating Mtb. However, TB is a complex and variegated disease, and human populations exhibit significant diversity in their immune responses to it, which presents a complicated landscape for HDTs to navigate. Nevertheless, we suggest that a detailed mechanistic understanding of drug action, together with careful selection of disease stage targets and dosing strategies may overcome such limitations and allow the development of HDTs as effective adjunctive treatment options for TB.
Keywords: drug; innate immunity; macrophage; tuberculosis.
© 2015 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
T. R. H., J. A. S., and D. K. have no conflicts of interest.
Figures
References
-
- Organization WH. WHO Annual Report. 2013. Global Tuberculosis Report 2013; pp. 1–289.
-
- Ma M, et al. Toll-like receptors, tumor necrosis factor–α, and interleukin-10 gene polymorphisms in risk of pulmonary tuberculosis and disease severity. HIM. 2010:1005–1010. - PubMed
-
- Swimm A, Bommarius B, Reeves P, Sherman M, Kalman D. Complex kinase requirements for EPEC pedestal formation. Nat Cell Biol. 2004;6:795. author reply 795–796. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
