

Identifying transcriptional cis-regulatory modules in animal genomes
- PMID: 25704908
- PMCID: PMC4339228
- DOI: 10.1002/wdev.168
Identifying transcriptional cis-regulatory modules in animal genomes
Abstract
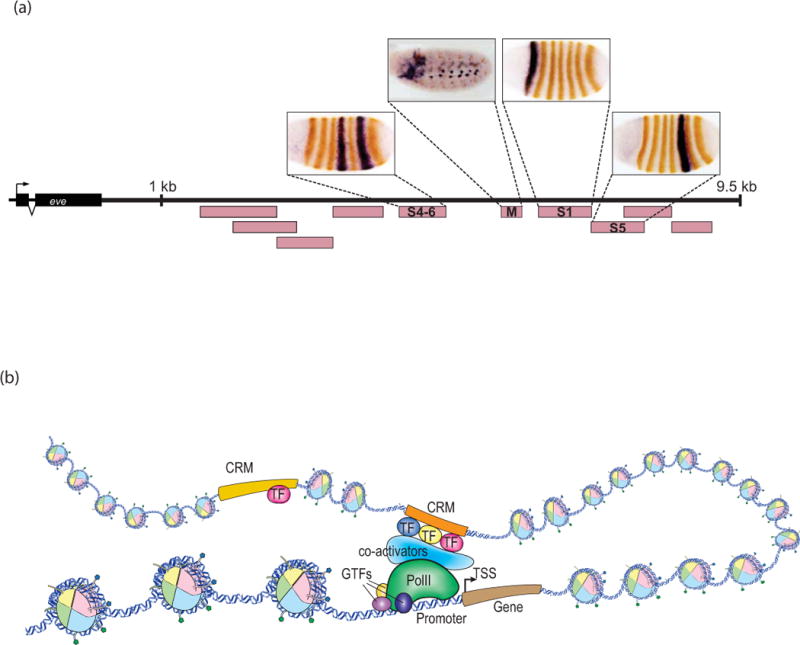
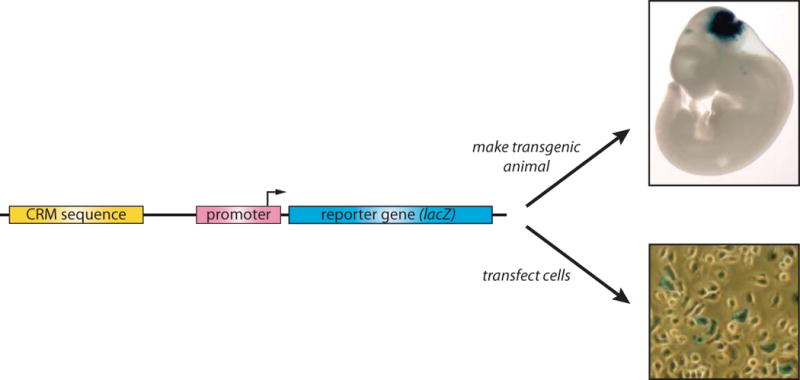
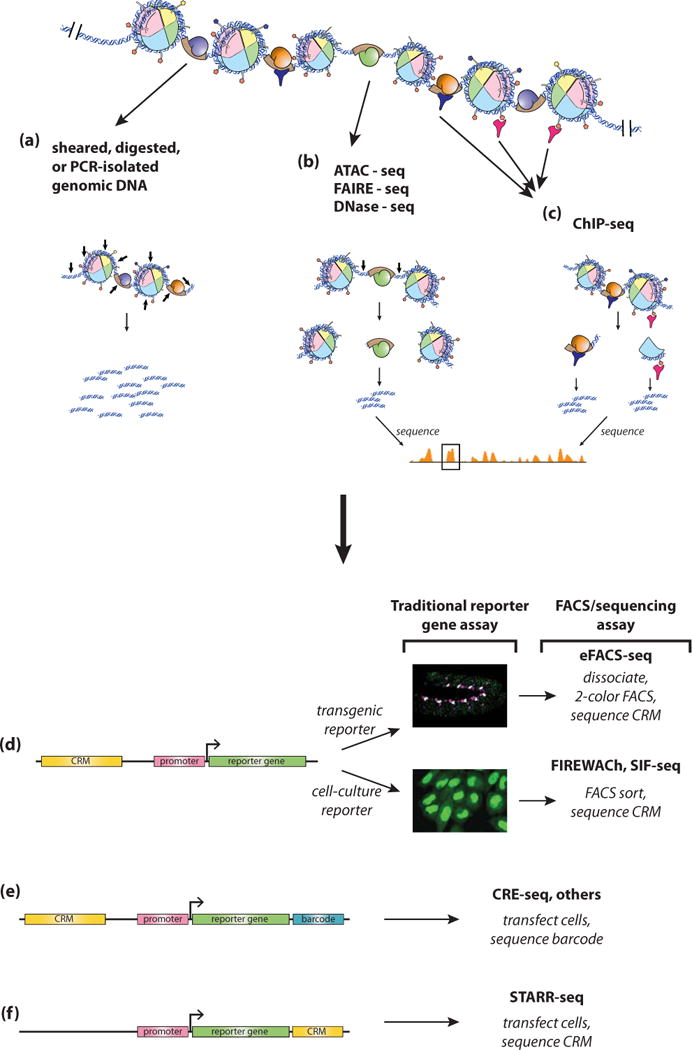
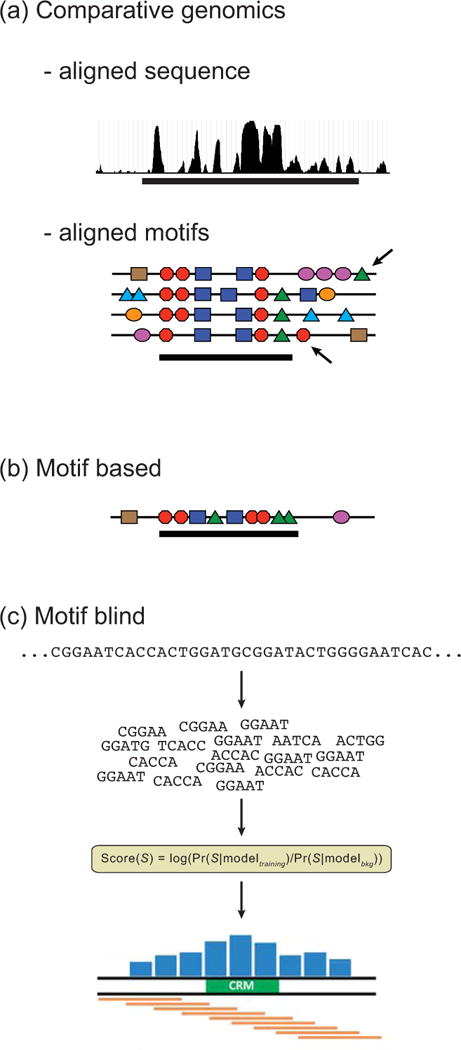
Gene expression is regulated through the activity of transcription factors (TFs) and chromatin-modifying proteins acting on specific DNA sequences, referred to as cis-regulatory elements. These include promoters, located at the transcription initiation sites of genes, and a variety of distal cis-regulatory modules (CRMs), the most common of which are transcriptional enhancers. Because regulated gene expression is fundamental to cell differentiation and acquisition of new cell fates, identifying, characterizing, and understanding the mechanisms of action of CRMs is critical for understanding development. CRM discovery has historically been challenging, as CRMs can be located far from the genes they regulate, have few readily identifiable sequence characteristics, and for many years were not amenable to high-throughput discovery methods. However, the recent availability of complete genome sequences and the development of next-generation sequencing methods have led to an explosion of both computational and empirical methods for CRM discovery in model and nonmodel organisms alike. Experimentally, CRMs can be identified through chromatin immunoprecipitation directed against TFs or histone post-translational modifications, identification of nucleosome-depleted 'open' chromatin regions, or sequencing-based high-throughput functional screening. Computational methods include comparative genomics, clustering of known or predicted TF-binding sites, and supervised machine-learning approaches trained on known CRMs. All of these methods have proven effective for CRM discovery, but each has its own considerations and limitations, and each is subject to a greater or lesser number of false-positive identifications. Experimental confirmation of predictions is essential, although shortcomings in current methods suggest that additional means of validation need to be developed. For further resources related to this article, please visit the WIREs website.
Conflict of interest: The authors have declared no conflicts of interest for this article.
© 2014 Wiley Periodicals, Inc.
Figures

References
RELATED ARTICLES
-
- Noordermeer D, Duboule D. Chromatin looping and organization at developmentally regulated gene loci. Wiley Interdiscip Rev Dev Biol. 2013;2(5):615–630. - PubMed
References
-
- Pick L, Heffer A. Hox gene evolution: multiple mechanisms contributing to evolutionary novelties. Ann N Y Acad Sci. 2012;1256:15–32. - PubMed
-
- Smith E, Shilatifard A. Enhancer biology and enhanceropathies. Nat Struct Mol Biol. 2014;21:210–219. - PubMed
-
- Maston GA, Landt SG, Snyder M, Green MR. Characterization of enhancer function from genome-wide analyses. Annu Rev Genomics Hum Genet. 2012;13:29–57. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
