

Nexavar/Stivarga and viagra interact to kill tumor cells
- PMID: 25704960
- PMCID: PMC4835179
- DOI: 10.1002/jcp.24961
Nexavar/Stivarga and viagra interact to kill tumor cells
Abstract
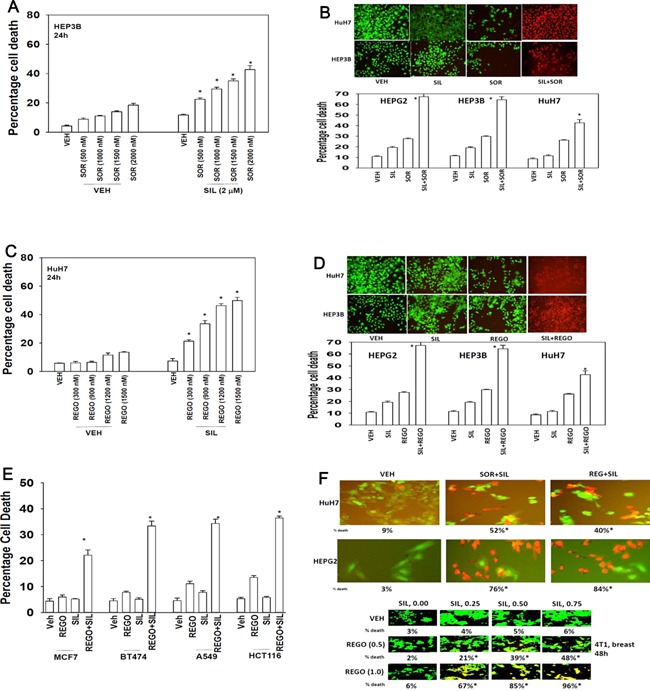
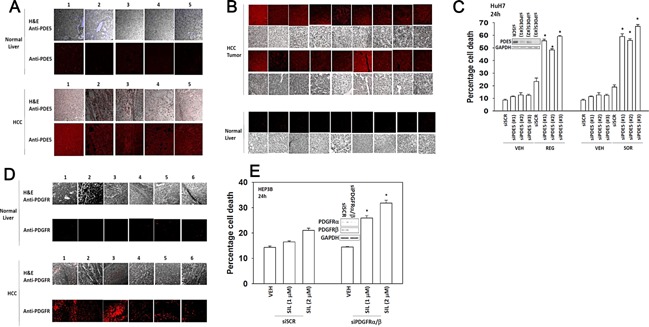
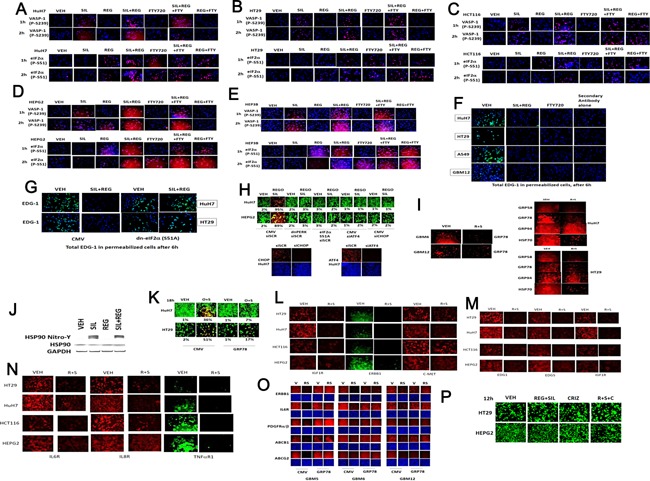
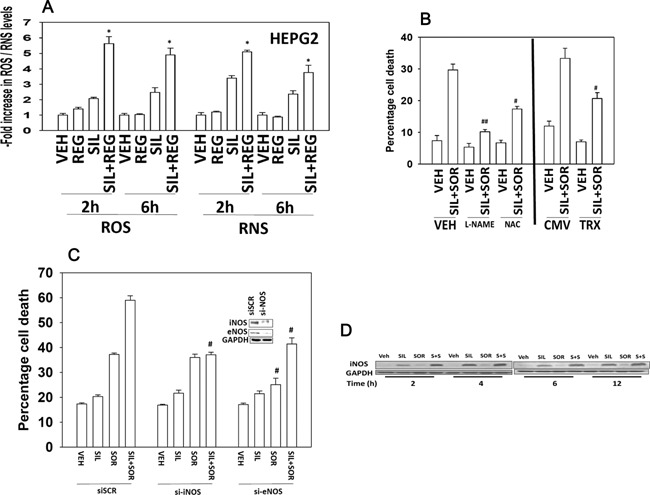
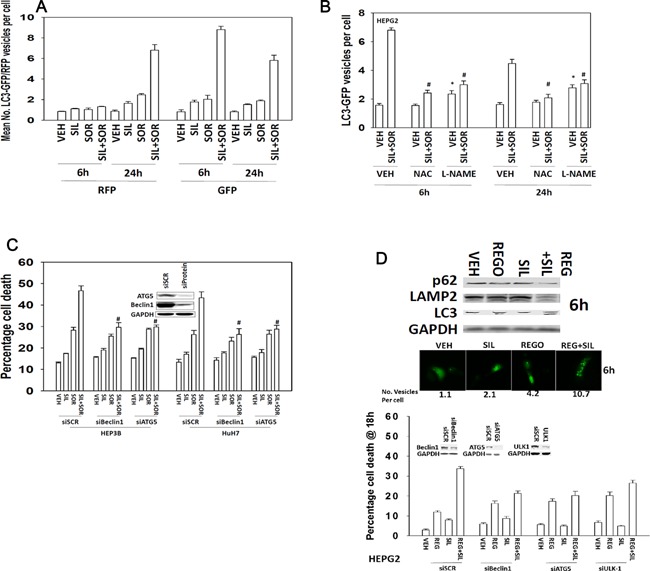
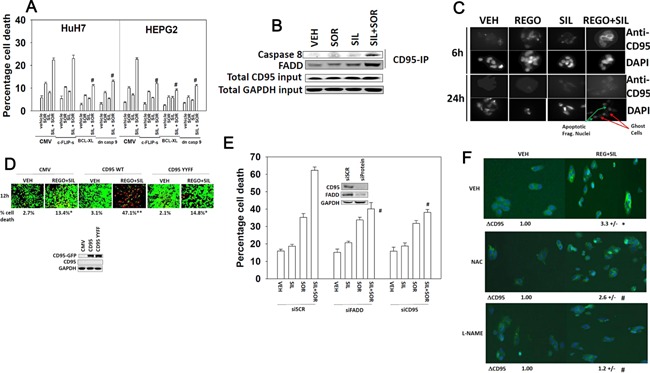
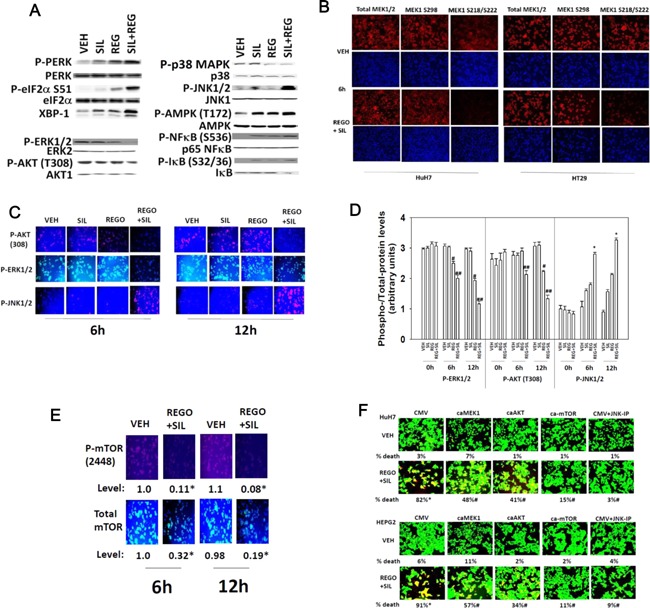
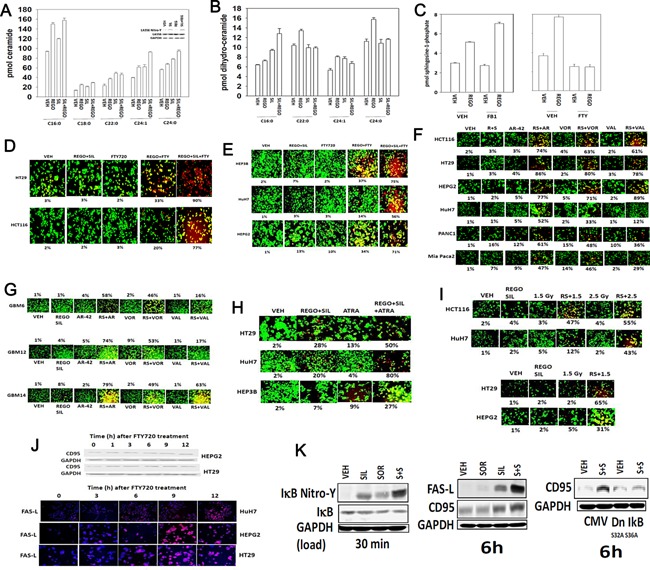
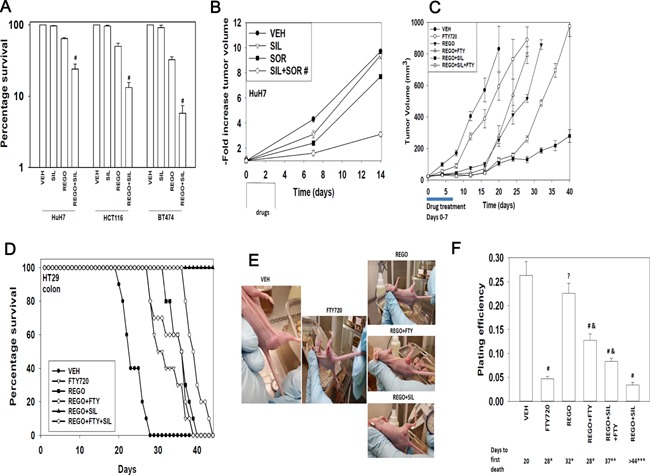
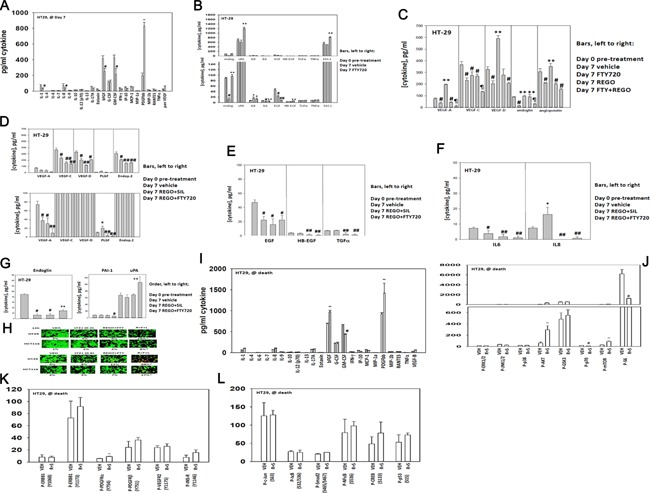
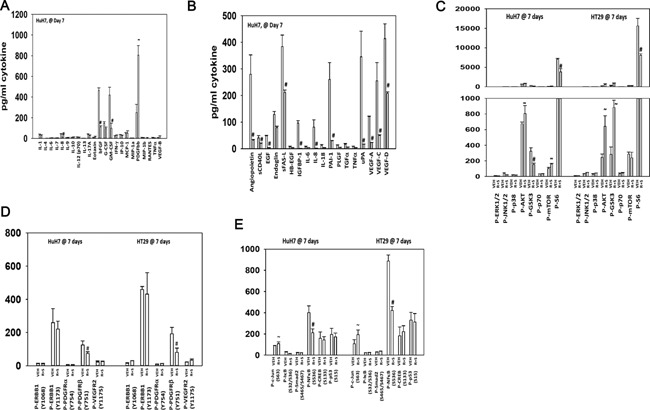
We determined whether the multi-kinase inhibitor sorafenib or its derivative regorafenib interacted with phosphodiesterase 5 (PDE5) inhibitors such as Viagra (sildenafil) to kill tumor cells. PDE5 and PDGFRα/β were over-expressed in liver tumors compared to normal liver tissue. In multiple cell types in vitro sorafenib/regorafenib and PDE5 inhibitors interacted in a greater than additive fashion to cause tumor cell death, regardless of whether cells were grown in 10 or 100% human serum. Knock down of PDE5 or of PDGFRα/β recapitulated the effects of the individual drugs. The drug combination increased ROS/RNS levels that were causal in cell killing. Inhibition of CD95/FADD/caspase 8 signaling suppressed drug combination toxicity. Knock down of ULK-1, Beclin1, or ATG5 suppressed drug combination lethality. The drug combination inactivated ERK, AKT, p70 S6K, and mTOR and activated JNK. The drug combination also reduced mTOR protein expression. Activation of ERK or AKT was modestly protective whereas re-expression of an activated mTOR protein or inhibition of JNK signaling almost abolished drug combination toxicity. Sildenafil and sorafenib/regorafenib interacted in vivo to suppress xenograft tumor growth using liver and colon cancer cells. From multiplex assays on tumor tissue and plasma, we discovered that increased FGF levels and ERBB1 and AKT phosphorylation were biomarkers that were directly associated with lower levels of cell killing by 'rafenib + sildenafil. Our data are now being translated into the clinic for further determination as to whether this drug combination is a useful anti-tumor therapy for solid tumor patients.
© 2015 Wiley Periodicals, Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
