

Spinal muscular atrophy: from tissue specificity to therapeutic strategies
- PMID: 25705387
- PMCID: PMC4311279
- DOI: 10.12703/P7-04
Spinal muscular atrophy: from tissue specificity to therapeutic strategies
Abstract
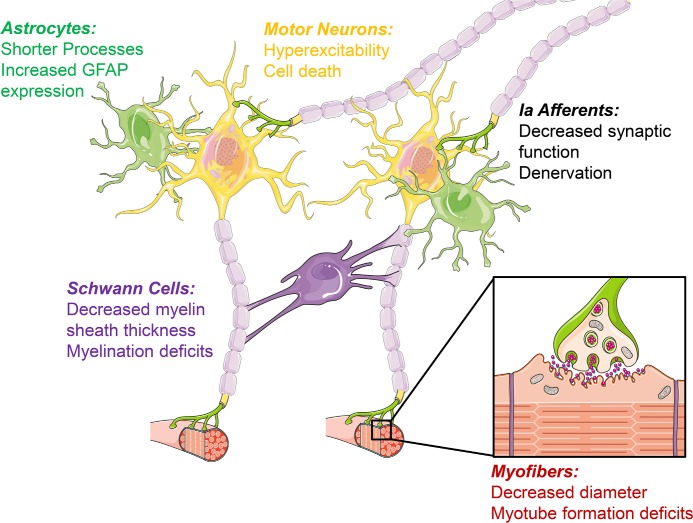
Spinal muscular atrophy (SMA) is the most frequent genetic cause of death in infants and toddlers. All cases of spinal muscular atrophy result from reductions in levels of the survival motor neuron (SMN) protein, and so SMN upregulation is a focus of many preclinical and clinical studies. We examine four issues that may be important in planning for therapeutic success. First, neuromuscular phenotypes in the SMNΔ7 mouse model closely match those in human patients but peripheral disease manifestations differ, suggesting that endpoints other than mouse lifespan may be more useful in predicting clinical outcome. Second, SMN plays important roles in multiple central and peripheral cell types, not just motor neurons, and it remains unclear which of these cell types need to be targeted therapeutically. Third, should SMN-restoration therapy not be effective in all patients, blocking molecular changes downstream of SMN reduction may confer significant benefit, making it important to evaluate therapeutic targets other than SMN. Lastly, for patients whose disease progression is slowed, but who retain significant motor dysfunction, additional approaches used to enhance regeneration of the neuromuscular system may be of value.
Figures

References
-
- Hoffmann J. Ueber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis. Deutsche Zeitschrift f Nervenheilkunde. 1893;3:427–70. doi: 10.1007/BF01668496. - DOI
-
- Hoffmann J. Dritter Beitrag zur Lehre von der hereditären progressiven spinalen Muskelatrophie im Kindesalter. Deutsche Zeitschrift f Nervenheilkunde. 1900;18:217–24. doi: 10.1007/BF01635796. - DOI
-
- Hoffmann J. Weiterer Beitrag zur Lehre von der hereditären progressiven spinalen Muskelatrophie im Kindesalter. Deutsche Zeitschrift f Nervenheilkunde. 1897;10:292–320. doi: 10.1007/BF01668174. - DOI
-
- Werdnig G. Zwei frühinfantile hereditäre Fälle von progressiver Muskelatrophie unter dem Bilde der Dystrophie, aber anf neurotischer Grundlage. Archiv f Psychiatrie. 1891;22:437–80.
-
- Beevor CE. A case of congenital spinal muscular atrophy (family type) and a case of hemorrhage into the spinal cord at birth, giving similar symptoms. Brain. 1902;25:85–108. doi: 10.1093/brain/25.1.85. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
