

How structural and physicochemical determinants shape sequence constraints in a functional enzyme
- PMID: 25706742
- PMCID: PMC4338278
- DOI: 10.1371/journal.pone.0118684
How structural and physicochemical determinants shape sequence constraints in a functional enzyme
Abstract
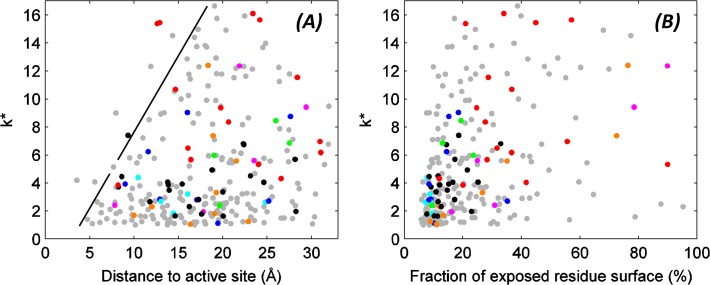
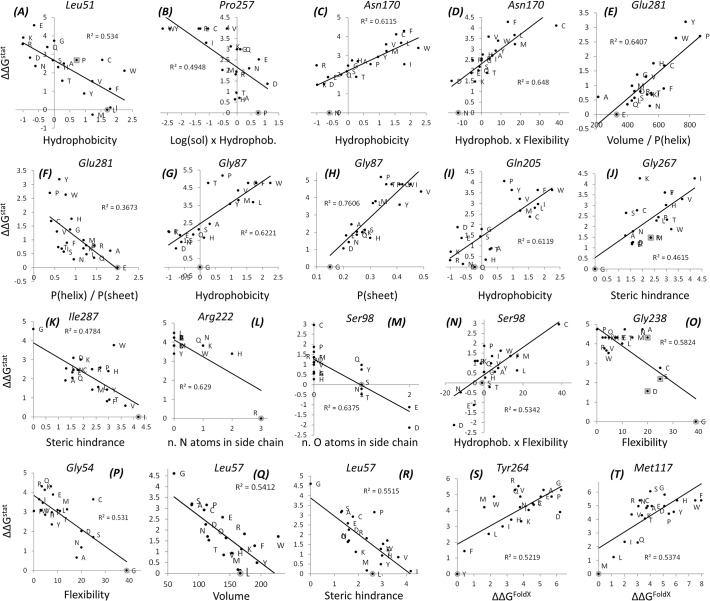
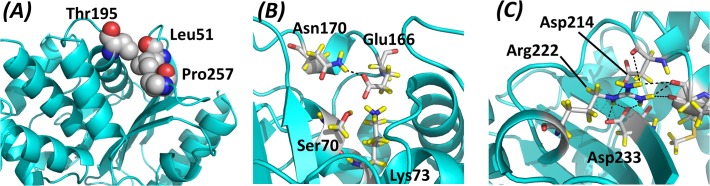
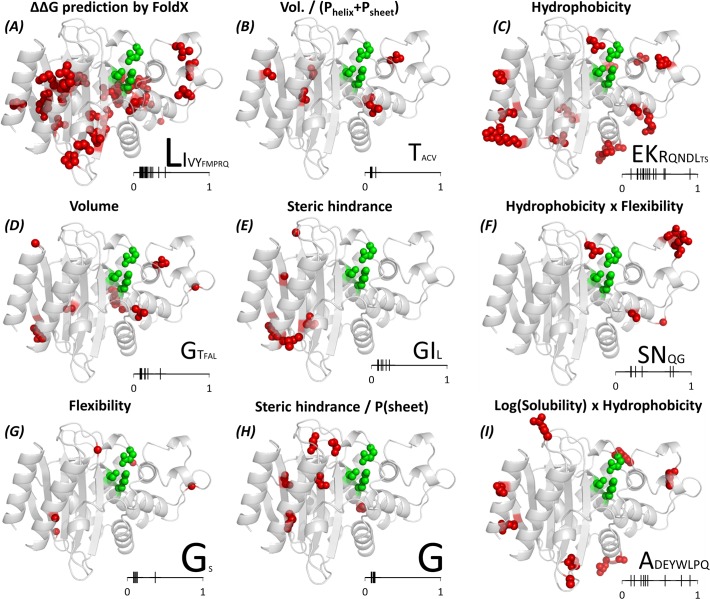
The need for interfacing structural biology and biophysics to molecular evolution is being increasingly recognized. One part of the big problem is to understand how physics and chemistry shape the sequence space available to functional proteins, while satisfying the needs of biology. Here we present a quantitative, structure-based analysis of a high-resolution map describing the tolerance to all substitutions in all positions of a functional enzyme, namely a TEM lactamase previously studied through deep sequencing of mutants growing in competition experiments with selection against ampicillin. Substitutions are rarely observed within 7 Å of the active site, a stringency that is relaxed slowly and extends up to 15-20 Å, with buried residues being especially sensitive. Substitution patterns in over one third of the residues can be quantitatively modeled by monotonic dependencies on amino acid descriptors and predictions of changes in folding stability. Amino acid volume and steric hindrance shape constraints on the protein core; hydrophobicity and solubility shape constraints on hydrophobic clusters underneath the surface, and on salt bridges and polar networks at the protein surface together with charge and hydrogen bonding capacity. Amino acid solubility, flexibility and conformational descriptors also provide additional constraints at many locations. These findings provide fundamental insights into the chemistry underlying protein evolution and design, by quantitating links between sequence and different protein traits, illuminating subtle and unexpected sequence-trait relationships and pinpointing what traits are sacrificed upon gain-of-function mutation.
Conflict of interest statement
Figures
References
-
- Aharoni A, Gaidukov L, Khersonsky O, McQ Gould S, Roodveldt C, et al. (2005) The “evolvability” of promiscuous protein functions. Nat Genet 37, 73–76 - PubMed
-
- Thorne JL (2007) Protein evolution constraints and model-based techniques to study them. Curr Opin Struct Biol 17, 337–341 - PubMed
-
- Fornasari MS, Parisi G, Echave J (2007) Quaternary structure constraints on evolutionary sequence divergence. Mol Biol Evol 24, 349–351 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
