

doi: 10.1038/nmeth.3286.
Epub 2015 Feb 23.
Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement
Affiliations
- PMID: 25707030
- PMCID: PMC4382417
- DOI: 10.1038/nmeth.3286
Item in Clipboard
Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement
Nat Methods.
2015 Apr.
Abstract
We describe a general approach for refining protein structure models on the basis of cryo-electron microscopy maps with near-atomic resolution. The method integrates Monte Carlo sampling with local density-guided optimization, Rosetta all-atom refinement and real-space B-factor fitting. In tests on experimental maps of three different systems with 4.5-Å resolution or better, the method consistently produced models with atomic-level accuracy largely independently of starting-model quality, and it outperformed the molecular dynamics-based MDFF method. Cross-validated model quality statistics correlated with model accuracy over the three test systems.
Figures
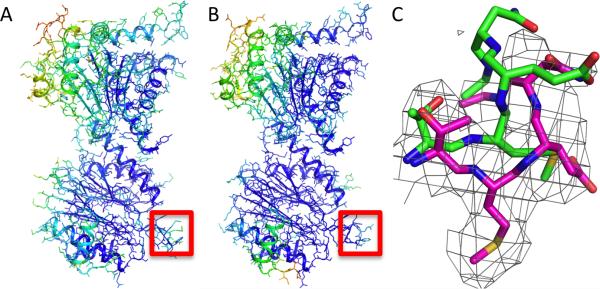
Refinement of 20S proteasome crystal structure into high-resolution cryoEM density. Atomic B-factors obtained from cryoEM model refinement correlate with the deposited X-ray B factors. (A,B) The crystal structure (PDB code: 1PMA) and the cryoEM model refined against the 3.3 Å map. The model is colored by the B-factor in the crystal structure (A), and by the Rosetta real-space B-factor fit to the cryoEM map (B). (C) An example of loop region that reconfigures in the cryoEM model: green, crystal structure; magenta, Rosetta refined model. The independent map density (not used in refinement) is shown.
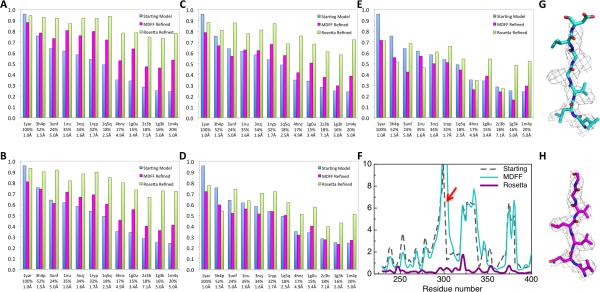
Dependence of model accuracy on starting model quality and map resolution. For a series of comparative models of 20S, Rosetta and MDFF refinement was initiated from comparative models based on templates indicated on the x-axis. The fraction of Cα atoms within 1 Å of the reference model is indicated on the y-axis for (blue) the starting comparative models, (magenta) the MDFF refined models, and (green) the Rosetta refined models. The templates are arranged from the best starting model to the worst based on the fraction of Cα atoms within 1 Å of the reference model. Sequence identity of the template and RMSD to the reference model is labeled under the PDB ID. Models were refined against 20S maps reconstructed using (A) 120,000 (B) 5,000 (C) 3,000 (D) 1,200 and (E) 1,000 particles, yielding 3.3, 4.1, 4.4, 5.0 and 6.0 Å resolution, respectively. (F) Deviations to the reference model (y-axis) from (black) the starting model based on 1g3k, (cyan) the MDFF refined model and (magenta) the Rosetta refined model for each residue (x-axis). Structure and electron density of the regions highlighted with red arrow is shown for (G) MDFF models and (H) Rosetta models.
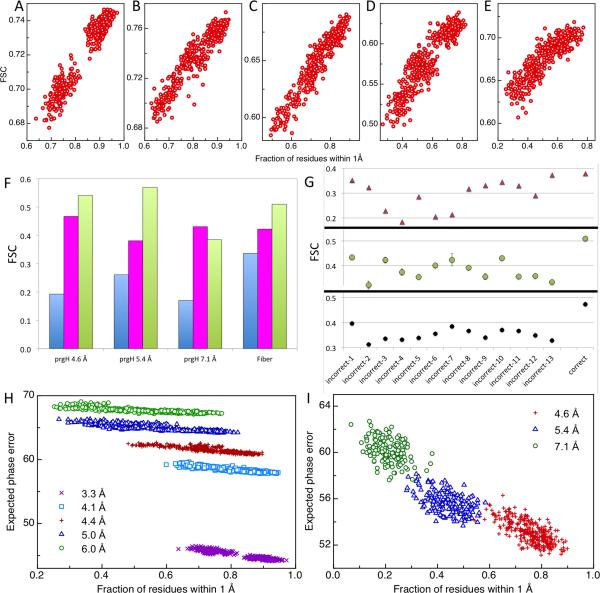
Model evaluation using independent maps. For each Rosetta-refined model of 20S at (A) 3.3 Å, (B) 4.1 Å, (C) 4.4 Å, (D) 5.0 Å, and (E) 6.0 Å resolution, the integrated FSC between model and testing map is plotted (y-axis) against the fraction of residues within 1 Å of the reference model (x-axis). More accurate models have higher independent map integrated FSC. (F) Evaluation of (blue) input models, (magenta) MDFF-, and (green) Rosetta-refined models for prgH and fiber based on the independent map integrated FSC. (G) Fiber models based on models (green); the MDFF models also identify the correct threading but with much weaker signal different sequence threading possibilities were refined in (magenta) MDFF and (green) Rosetta. The correct threading is distinguished by the highest integrated FSC in the Rosetta refined (magenta). The integrated FSC between Rosetta models refined in this study and a higher resolution density map available more recently (black) validates the threading identified using Rosetta and the lower resolution map (green). (H,I) Expected phase error (y-axis) correlates with the accuracies of refined models (x-axis). Refinement was carried out with reconstructed maps of 20S proteasome (H) at (magenta) 3.3Å, (cyan) 4.1 Å, (red) 4.4 Å, (blue) 5.0 Å and (green) 6.0 Å, and with maps of prgH (I) at (red) 4.6 Å, (blue) 5.4 Å and (green) 7.1 Å. The expected phase error tracks absolute model quality better than the integrated FSC (Supplemental Fig. 3).
Similar articles
-
De novo protein structure determination from near-atomic-resolution cryo-EM maps.Nat Methods. 2015 Apr;12(4):335-8. doi: 10.1038/nmeth.3287. Epub 2015 Feb 23. Nat Methods. 2015. PMID: 25707029 Free PMC article.
-
Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement.J Chem Inf Model. 2020 May 26;60(5):2522-2532. doi: 10.1021/acs.jcim.9b00932. Epub 2019 Dec 24. J Chem Inf Model. 2020. PMID: 31872764 Free PMC article.
-
A New Protocol for Atomic-Level Protein Structure Modeling and Refinement Using Low-to-Medium Resolution Cryo-EM Density Maps.J Mol Biol. 2020 Sep 4;432(19):5365-5377. doi: 10.1016/j.jmb.2020.07.027. Epub 2020 Aug 6. J Mol Biol. 2020. PMID: 32771523 Free PMC article.
-
StarMap: a user-friendly workflow for Rosetta-driven molecular structure refinement.Nat Protoc. 2023 Jan;18(1):239-264. doi: 10.1038/s41596-022-00757-9. Epub 2022 Nov 2. Nat Protoc. 2023. PMID: 36323866 Review.
-
Simulation-Based Methods for Model Building and Refinement in Cryoelectron Microscopy.J Chem Inf Model. 2020 May 26;60(5):2470-2483. doi: 10.1021/acs.jcim.0c00087. Epub 2020 Mar 31. J Chem Inf Model. 2020. PMID: 32202798 Free PMC article. Review.
Cited by
-
N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2.Cell. 2021 Apr 29;184(9):2332-2347.e16. doi: 10.1016/j.cell.2021.03.028. Epub 2021 Mar 16. Cell. 2021. PMID: 33761326 Free PMC article.
-
Automatically Fixing Errors in Glycoprotein Structures with Rosetta.Structure. 2019 Jan 2;27(1):134-139.e3. doi: 10.1016/j.str.2018.09.006. Epub 2018 Oct 18. Structure. 2019. PMID: 30344107 Free PMC article.
-
Structural Basis of Pan-Ebolavirus Neutralization by an Antibody Targeting the Glycoprotein Fusion Loop.Cell Rep. 2018 Sep 4;24(10):2723-2732.e4. doi: 10.1016/j.celrep.2018.08.009. Cell Rep. 2018. PMID: 30184505 Free PMC article.
-
Structure of a Tc holotoxin pore provides insights into the translocation mechanism.Proc Natl Acad Sci U S A. 2019 Nov 12;116(46):23083-23090. doi: 10.1073/pnas.1909821116. Epub 2019 Oct 30. Proc Natl Acad Sci U S A. 2019. PMID: 31666324 Free PMC article.
-
Role of Computational Methods in Going beyond X-ray Crystallography to Explore Protein Structure and Dynamics.Int J Mol Sci. 2018 Oct 30;19(11):3401. doi: 10.3390/ijms19113401. Int J Mol Sci. 2018. PMID: 30380757 Free PMC article. Review.
References
-
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
