

Hypothalamic POMC neurons promote cannabinoid-induced feeding
- PMID: 25707796
- PMCID: PMC4496586
- DOI: 10.1038/nature14260
Hypothalamic POMC neurons promote cannabinoid-induced feeding
Abstract
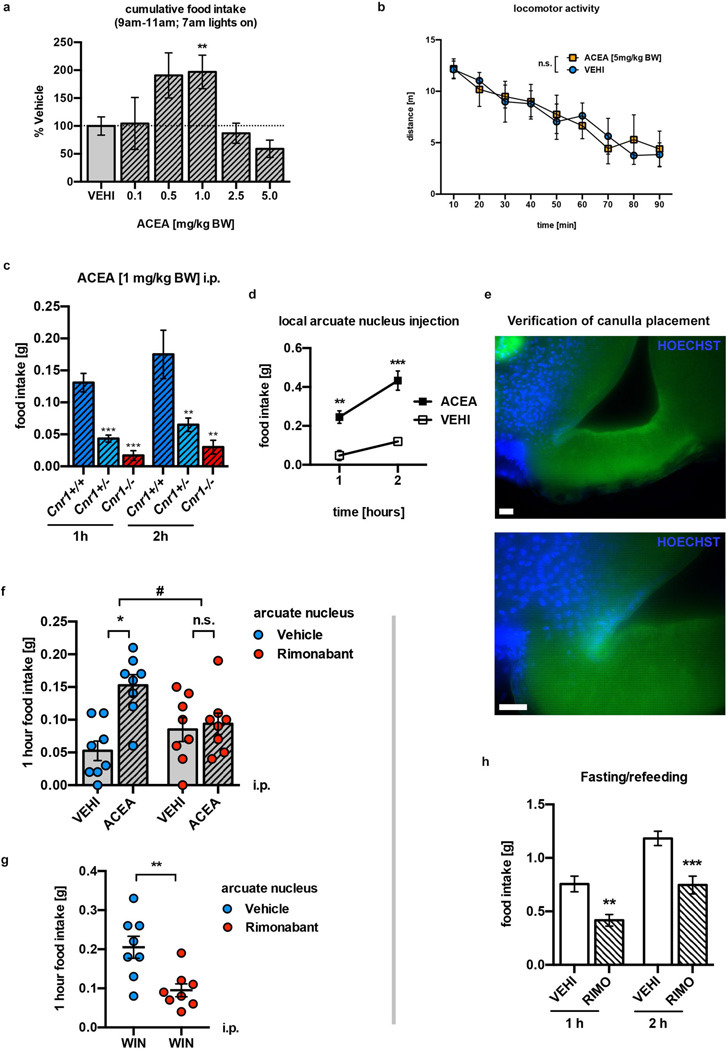
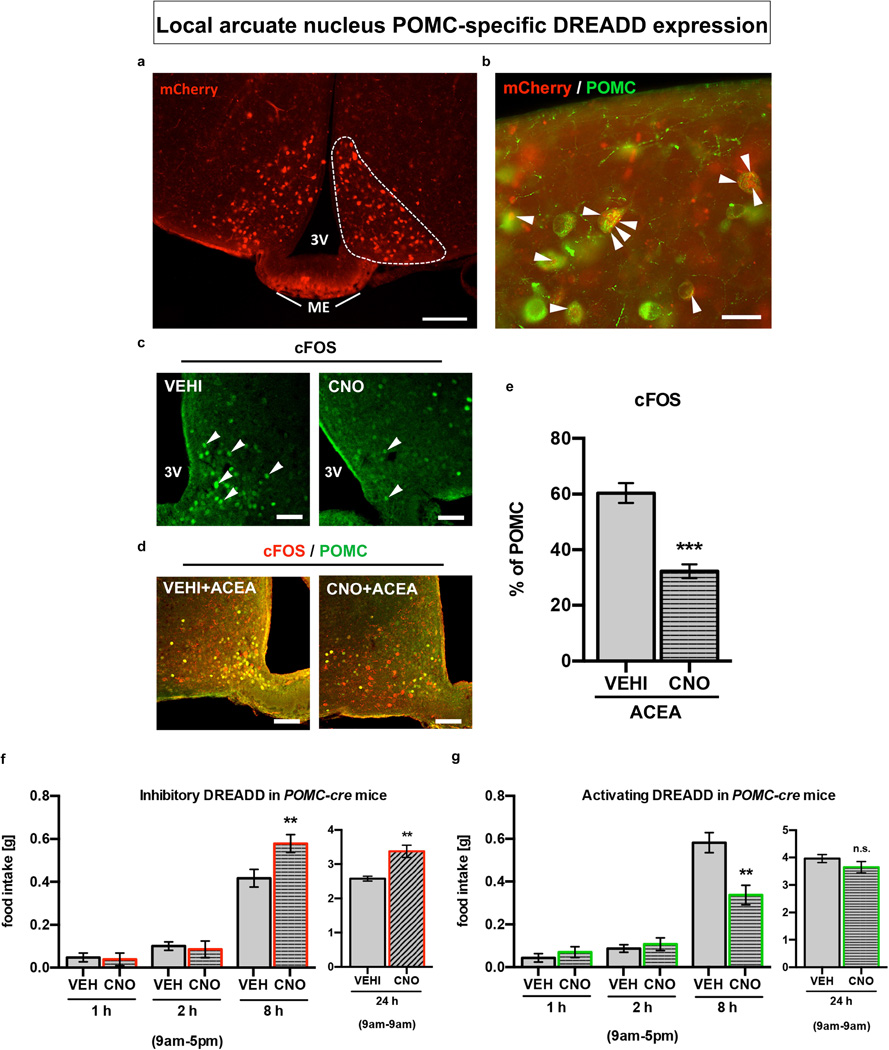
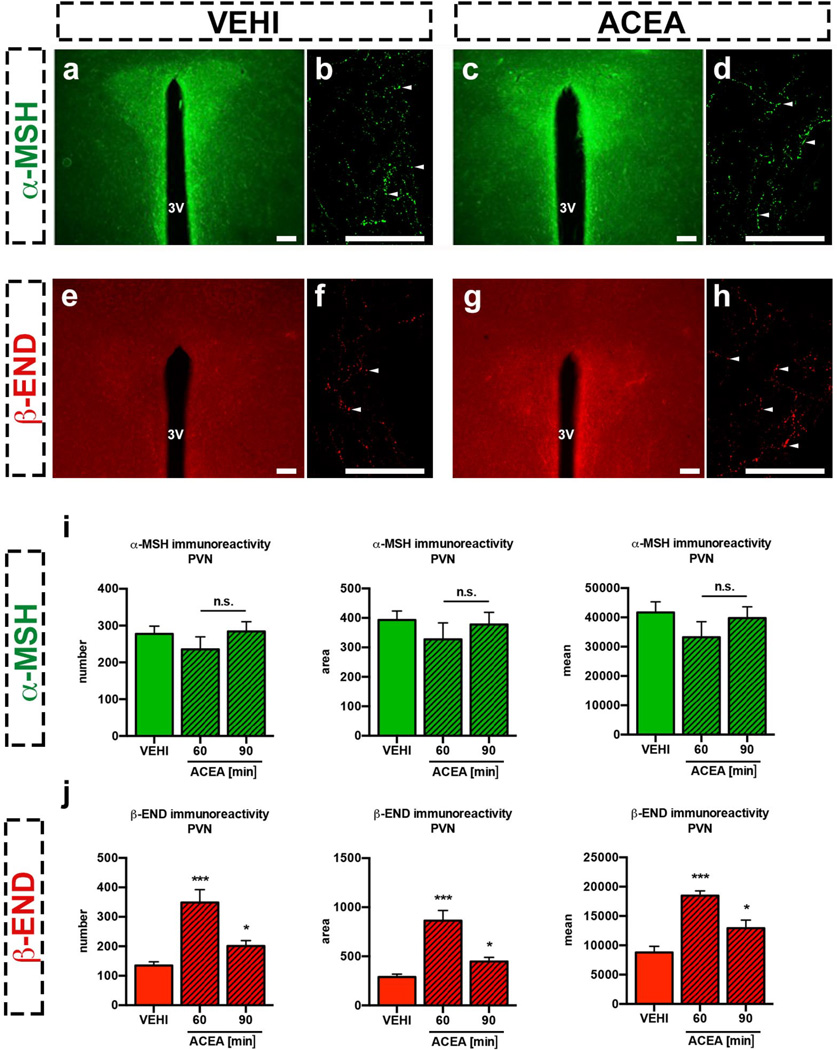
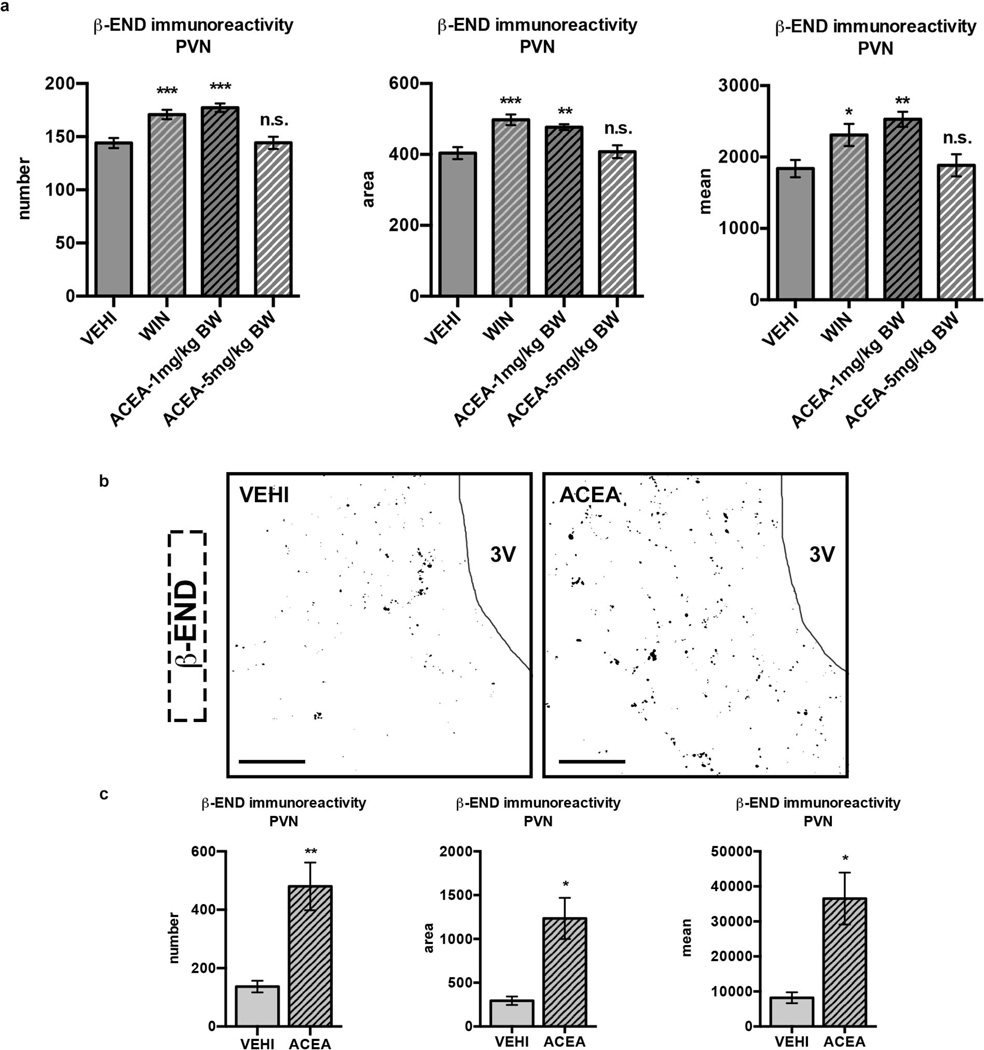
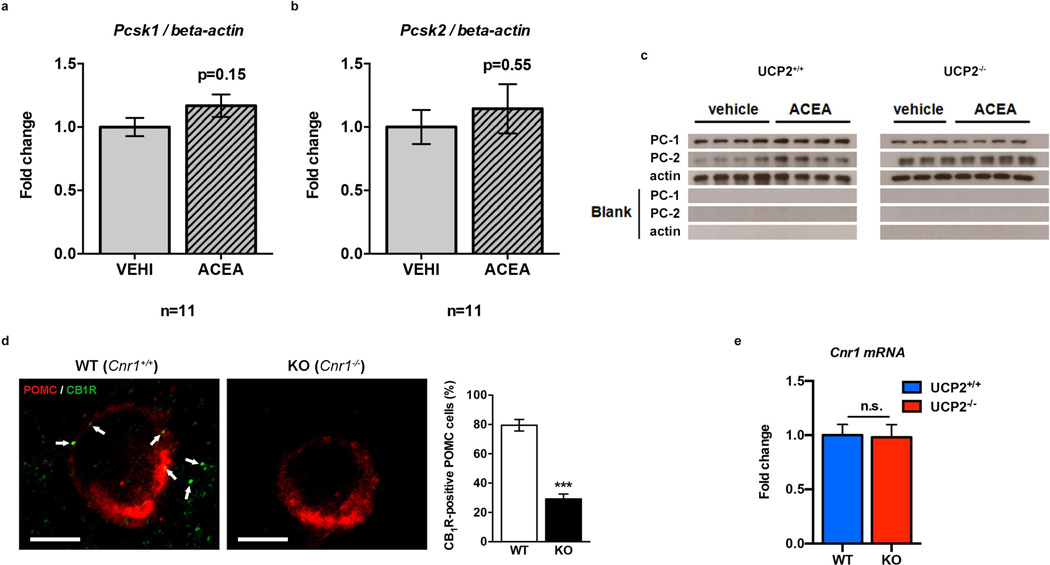
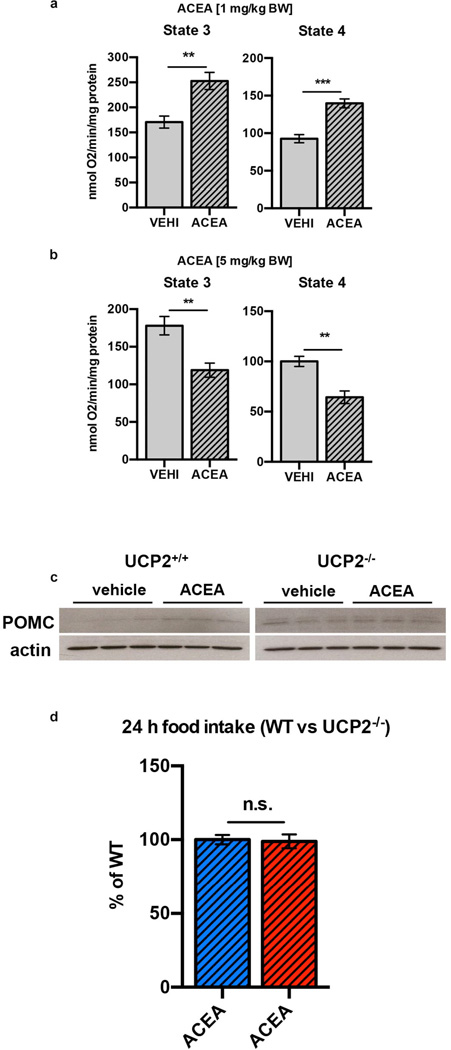
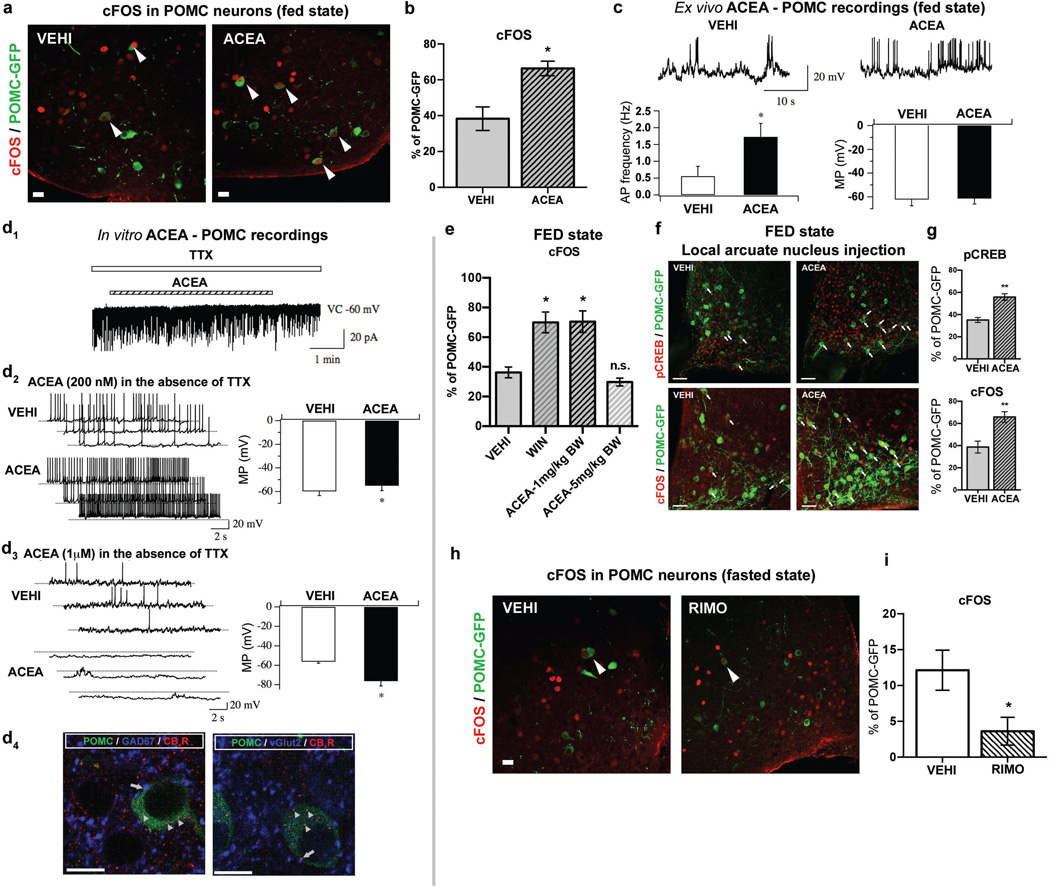
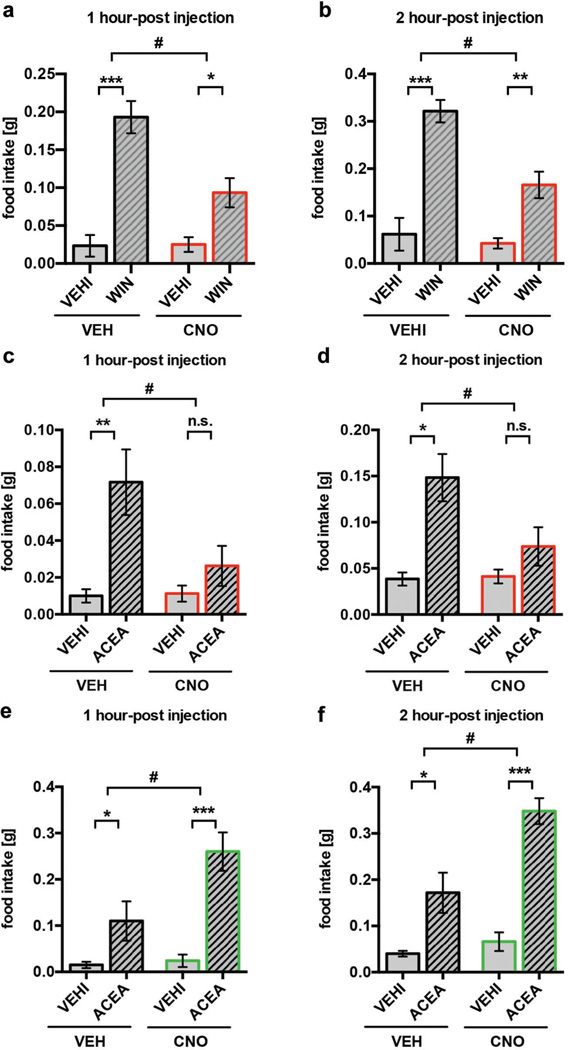
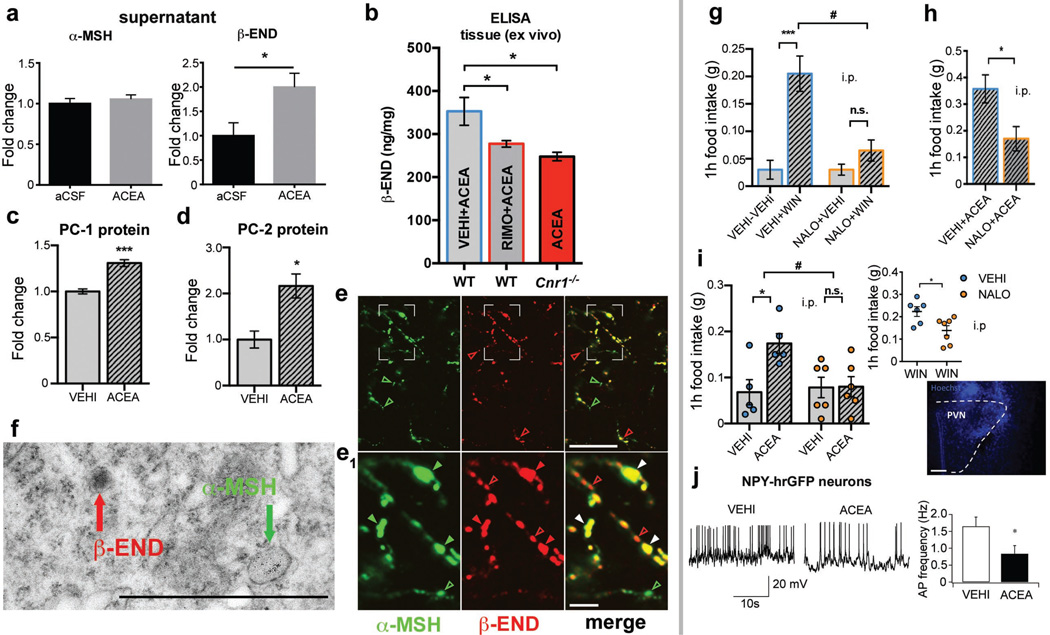
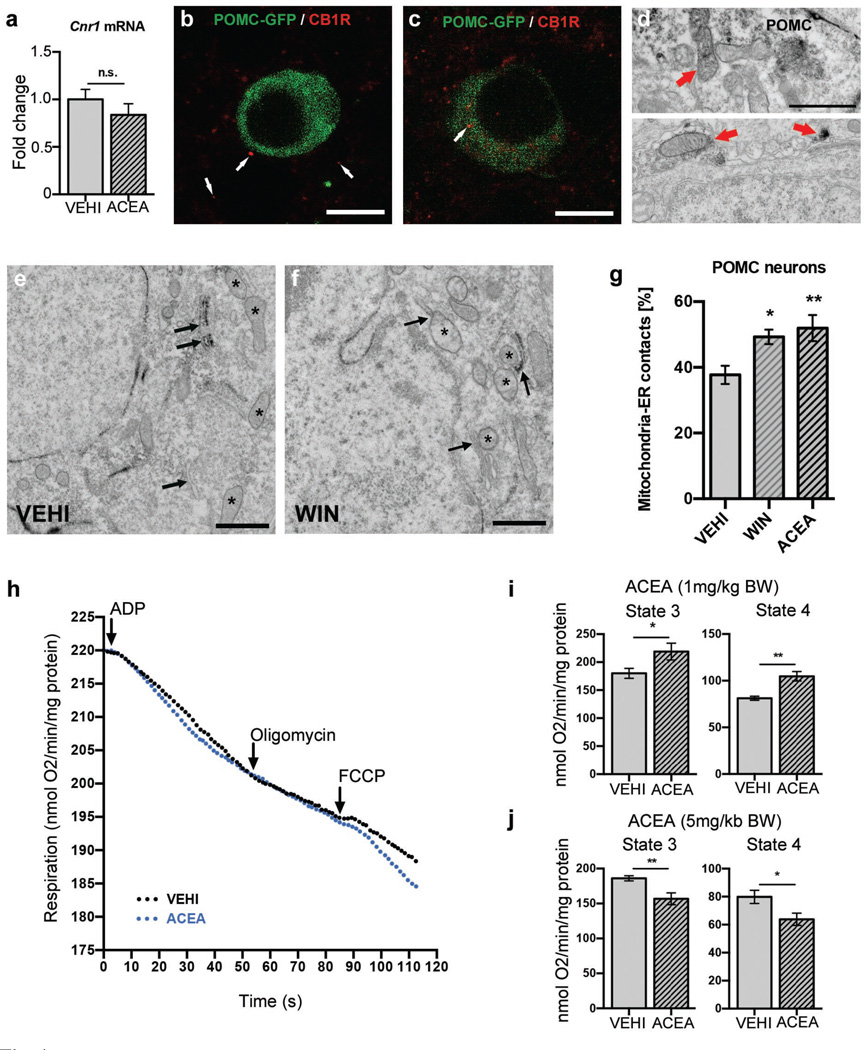
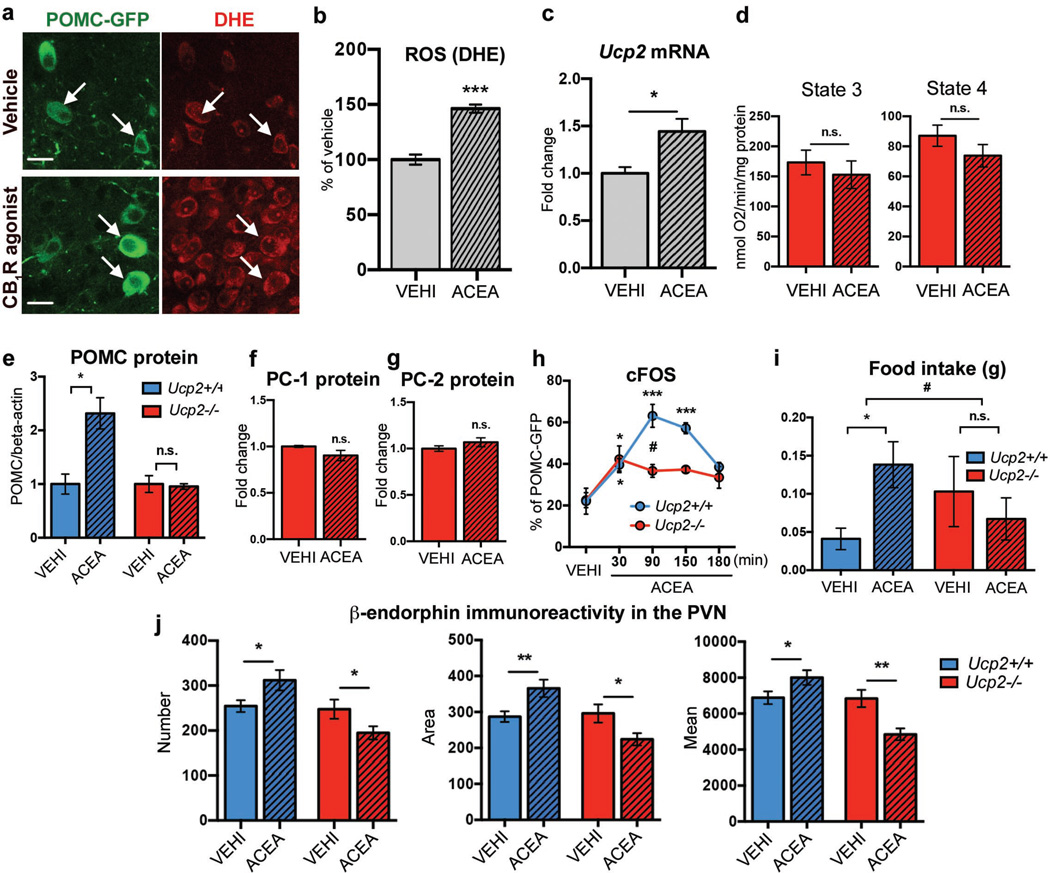
Hypothalamic pro-opiomelanocortin (POMC) neurons promote satiety. Cannabinoid receptor 1 (CB1R) is critical for the central regulation of food intake. Here we test whether CB1R-controlled feeding in sated mice is paralleled by decreased activity of POMC neurons. We show that chemical promotion of CB1R activity increases feeding, and notably, CB1R activation also promotes neuronal activity of POMC cells. This paradoxical increase in POMC activity was crucial for CB1R-induced feeding, because designer-receptors-exclusively-activated-by-designer-drugs (DREADD)-mediated inhibition of POMC neurons diminishes, whereas DREADD-mediated activation of POMC neurons enhances CB1R-driven feeding. The Pomc gene encodes both the anorexigenic peptide α-melanocyte-stimulating hormone, and the opioid peptide β-endorphin. CB1R activation selectively increases β-endorphin but not α-melanocyte-stimulating hormone release in the hypothalamus, and systemic or hypothalamic administration of the opioid receptor antagonist naloxone blocks acute CB1R-induced feeding. These processes involve mitochondrial adaptations that, when blocked, abolish CB1R-induced cellular responses and feeding. Together, these results uncover a previously unsuspected role of POMC neurons in the promotion of feeding by cannabinoids.
Figures
Comment in
-
Neuroscience: a cellular basis for the munchies.Nature. 2015 Mar 5;519(7541):38-40. doi: 10.1038/nature14206. Epub 2015 Feb 18. Nature. 2015. PMID: 25707800 No abstract available.
-
Neuropharmacology: Driving the urge to eat.Nat Rev Neurosci. 2015 Apr;16(4):185. doi: 10.1038/nrn3940. Epub 2015 Mar 11. Nat Rev Neurosci. 2015. PMID: 25757559 No abstract available.
References
-
- Dietrich MO, Horvath TL. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends in neurosciences. 2013;36:65–73. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
