

A linear time algorithm for detecting long genomic regions enriched with a specific combination of epigenetic states
- PMID: 25708947
- PMCID: PMC4331722
- DOI: 10.1186/1471-2164-16-S2-S8
A linear time algorithm for detecting long genomic regions enriched with a specific combination of epigenetic states
Abstract
Background: Epigenetic modifications are essential for controlling gene expression. Recent studies have shown that not only single epigenetic modifications but also combinations of multiple epigenetic modifications play vital roles in gene regulation. A striking example is the long hypomethylated regions enriched with modified H3K27me3 (called, "K27HMD" regions), which are exposed to suppress the expression of key developmental genes relevant to cellular development and differentiation during embryonic stages in vertebrates. It is thus a biologically important issue to develop an effective optimization algorithm for detecting long DNA regions (e.g., >4 kbp in size) that harbor a specific combination of epigenetic modifications (e.g., K27HMD regions). However, to date, optimization algorithms for these purposes have received little attention, and available methods are still heuristic and ad hoc.
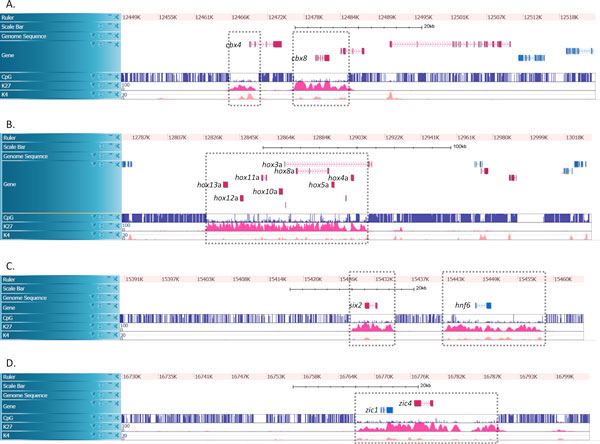
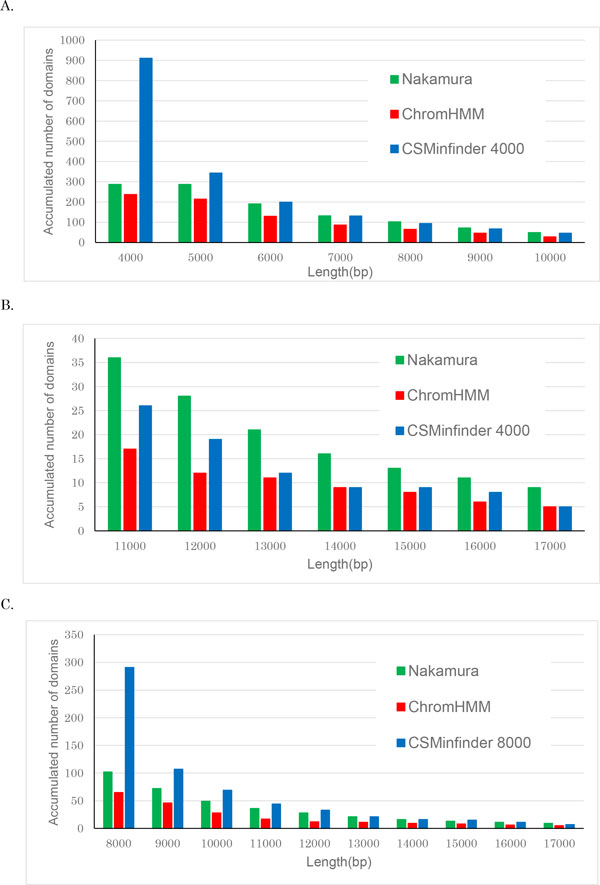
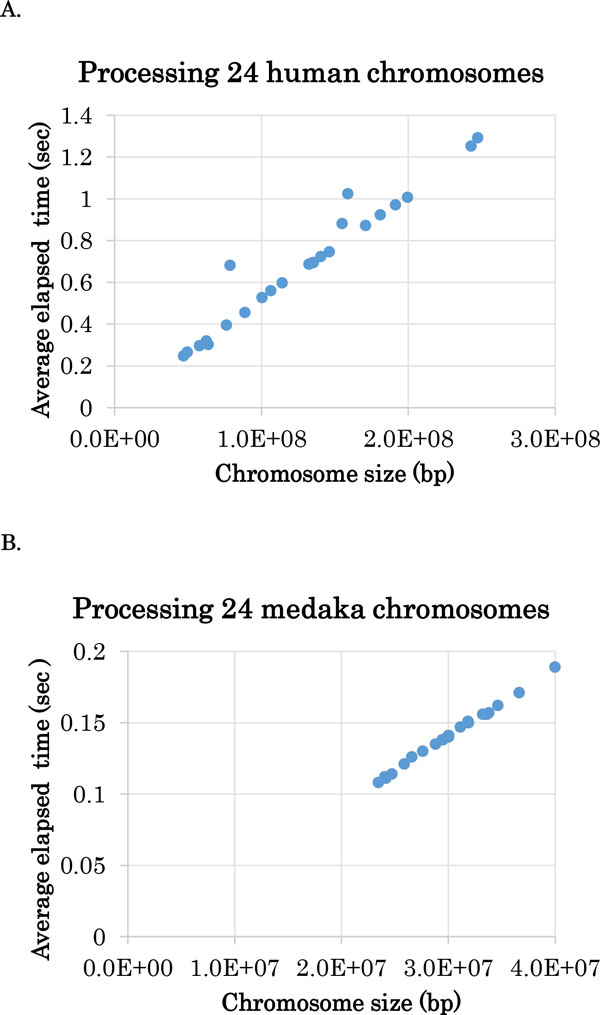
Results: In this paper, we propose a linear time algorithm for calculating a set of non-overlapping regions that maximizes the sum of similarities between the vector of focal epigenetic states and the vectors of raw epigenetic states at DNA positions in the set of regions. The average elapsed time to process the epigenetic data of any of human chromosomes was less than 2 seconds on an Intel Xeon CPU. To demonstrate the effectiveness of the algorithm, we estimated large K27HMD regions in the medaka and human genomes using our method, ChromHMM, and a heuristic method.
Conclusions: We confirmed that the advantages of our method over those of the two other methods. Our method is flexible enough to handle other types of epigenetic combinations. The program that implements the method is called "CSMinfinder" and is made available at: http://mlab.cb.k.u-tokyo.ac.jp/~ichikawa/Segmentation/
Figures



Similar articles
-
Large hypomethylated domains serve as strong repressive machinery for key developmental genes in vertebrates.Development. 2014 Jul;141(13):2568-80. doi: 10.1242/dev.108548. Epub 2014 Jun 12. Development. 2014. PMID: 24924192
-
Unlinking the methylome pattern from nucleotide sequence, revealed by large-scale in vivo genome engineering and methylome editing in medaka fish.PLoS Genet. 2017 Dec 21;13(12):e1007123. doi: 10.1371/journal.pgen.1007123. eCollection 2017 Dec. PLoS Genet. 2017. PMID: 29267279 Free PMC article.
-
Epigenetic signatures and temporal expression of lineage-specific genes in hESCs during differentiation to hepatocytes in vitro.Hum Mol Genet. 2011 Feb 1;20(3):401-12. doi: 10.1093/hmg/ddq476. Epub 2010 Nov 8. Hum Mol Genet. 2011. PMID: 21059703
-
An integrated workflow for DNA methylation analysis.J Genet Genomics. 2013 May 20;40(5):249-60. doi: 10.1016/j.jgg.2013.03.010. Epub 2013 Mar 30. J Genet Genomics. 2013. PMID: 23706300 Review.
-
Next generation sequencing in epigenetics: insights and challenges.Semin Cell Dev Biol. 2012 Apr;23(2):192-9. doi: 10.1016/j.semcdb.2011.10.010. Epub 2011 Oct 19. Semin Cell Dev Biol. 2012. PMID: 22027613 Review.
References
-
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
