

Rare variant association studies: considerations, challenges and opportunities
- PMID: 25709717
- PMCID: PMC4337325
- DOI: 10.1186/s13073-015-0138-2
Rare variant association studies: considerations, challenges and opportunities
Abstract
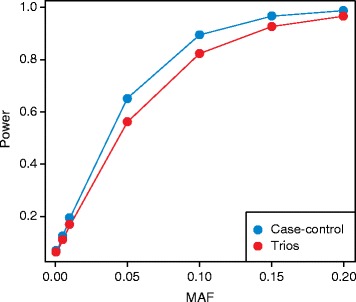
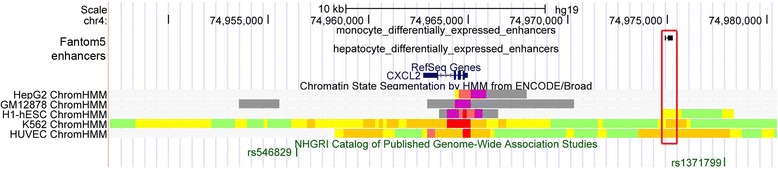
Genome-wide association studies (GWASs) have successfully uncovered thousands of robust associations between common variants and complex traits and diseases. Despite these successes, much of the heritability of these traits remains unexplained. Because low-frequency and rare variants are not tagged by conventional genome-wide genotyping arrays, they may represent an important and understudied component of complex trait genetics. In contrast to common variant GWASs, there are many different types of study designs, assays and analytic techniques that can be utilized for rare variant association studies (RVASs). In this review, we briefly present the different technologies available to identify rare genetic variants, including novel exome arrays. We also compare the different study designs for RVASs and argue that the best design will likely be phenotype-dependent. We discuss the main analytical issues relevant to RVASs, including the different statistical methods that can be used to test genetic associations with rare variants and the various bioinformatic approaches to predicting in silico biological functions for variants. Finally, we describe recent rare variant association findings, highlighting the unexpected conclusion that most rare variants have modest-to-small effect sizes on phenotypic variation. This observation has major implications for our understanding of the genetic architecture of complex traits in the context of the unexplained heritability challenge.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
