

X-linked acrogigantism syndrome: clinical profile and therapeutic responses
- PMID: 25712922
- PMCID: PMC4433400
- DOI: 10.1530/ERC-15-0038
X-linked acrogigantism syndrome: clinical profile and therapeutic responses
Abstract
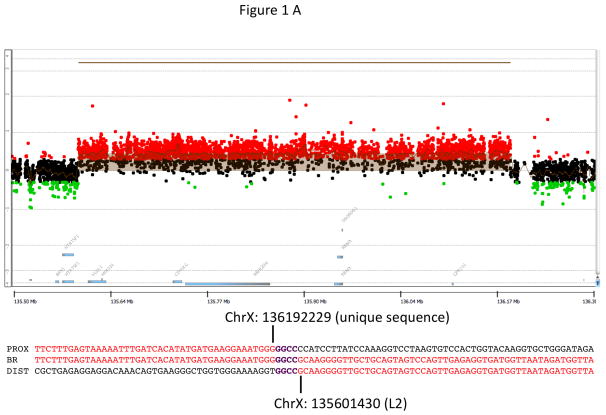
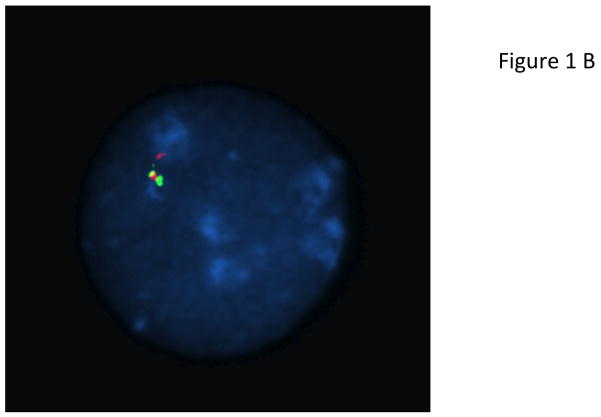
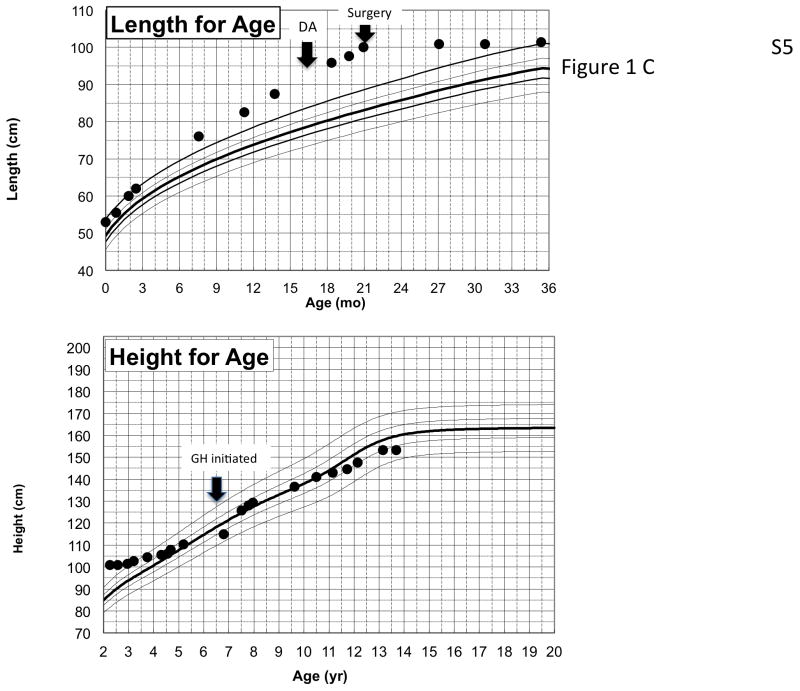
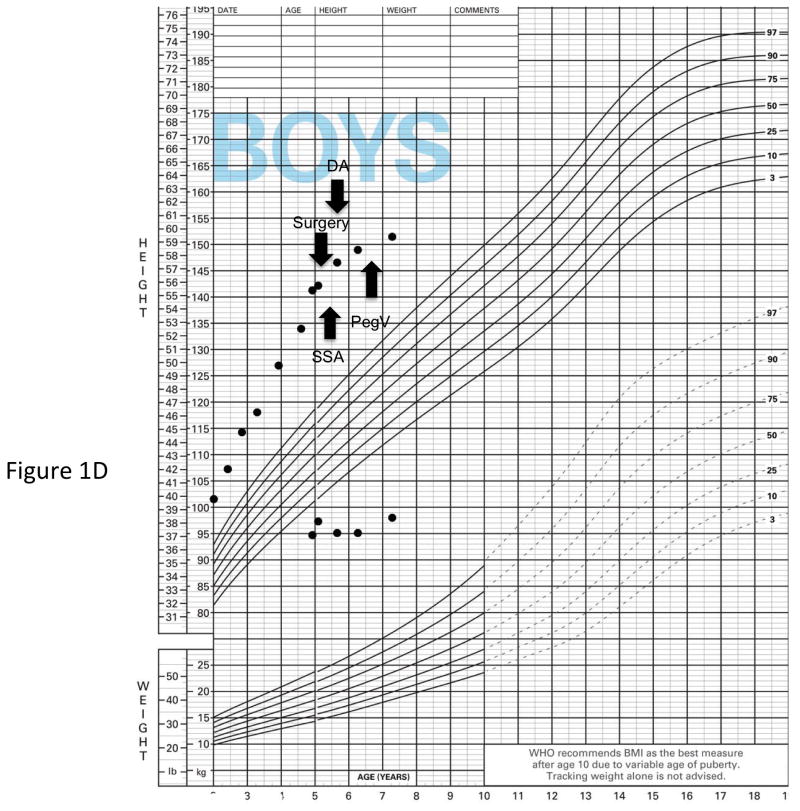
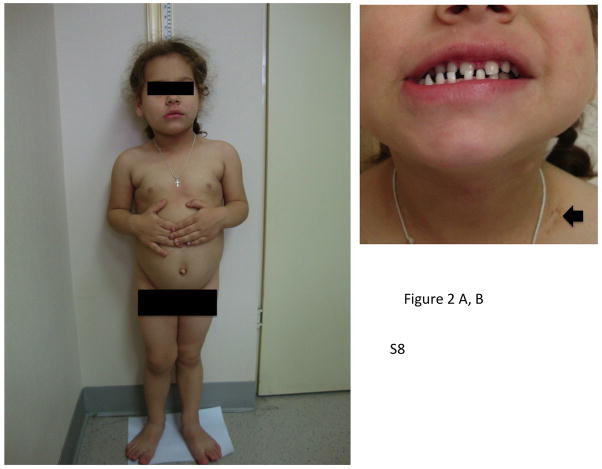
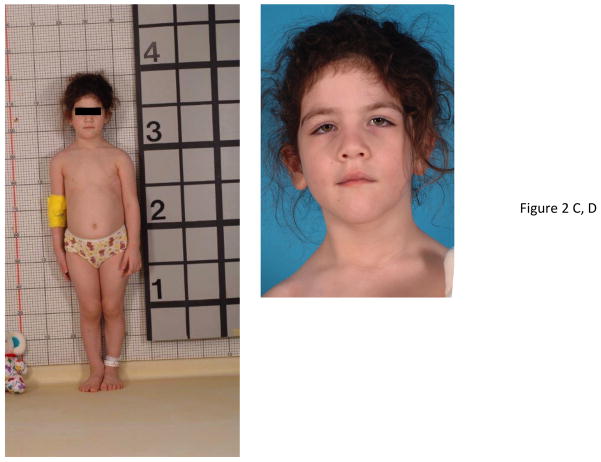
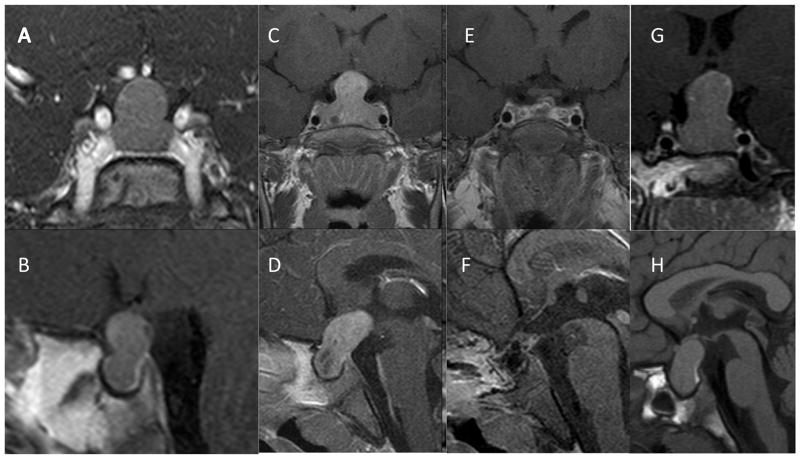
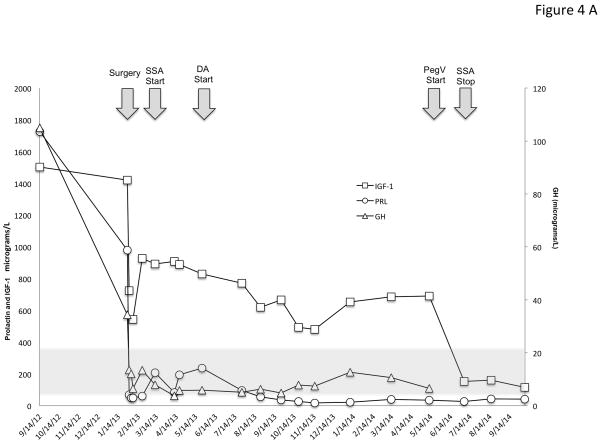
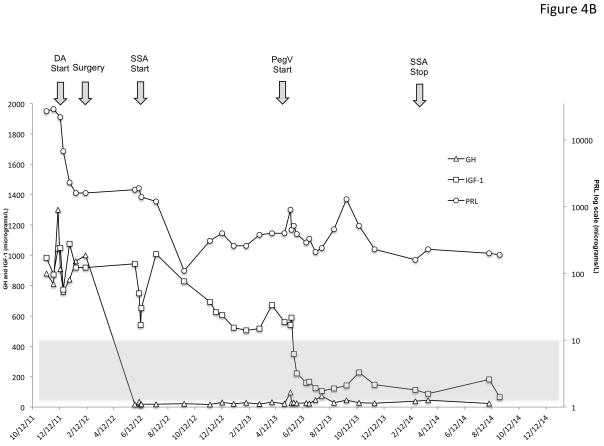
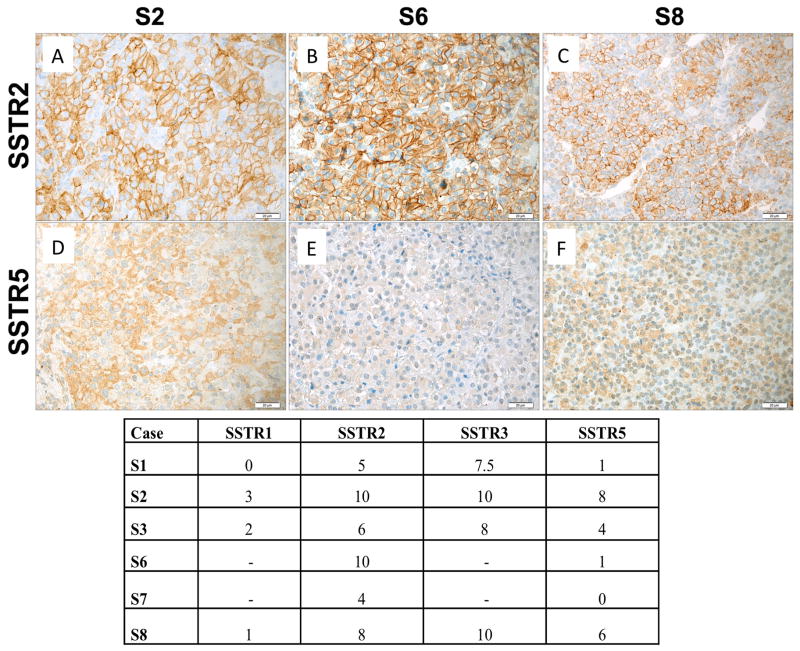
X-linked acrogigantism (X-LAG) is a new syndrome of pituitary gigantism, caused by microduplications on chromosome Xq26.3, encompassing the gene GPR101, which is highly upregulated in pituitary tumors. We conducted this study to explore the clinical, radiological, and hormonal phenotype and responses to therapy in patients with X-LAG syndrome. The study included 18 patients (13 sporadic) with X-LAG and microduplication of chromosome Xq26.3. All sporadic cases had unique duplications and the inheritance pattern in two families was dominant, with all Xq26.3 duplication carriers being affected. Patients began to grow rapidly as early as 2-3 months of age (median 12 months). At diagnosis (median delay 27 months), patients had a median height and weight standard deviation scores (SDS) of >+3.9 SDS. Apart from the increased overall body size, the children had acromegalic symptoms including acral enlargement and facial coarsening. More than a third of cases had increased appetite. Patients had marked hypersecretion of GH/IGF1 and usually prolactin, due to a pituitary macroadenoma or hyperplasia. Primary neurosurgical control was achieved with extensive anterior pituitary resection, but postoperative hypopituitarism was frequent. Control with somatostatin analogs was not readily achieved despite moderate to high levels of expression of somatostatin receptor subtype-2 in tumor tissue. Postoperative use of adjuvant pegvisomant resulted in control of IGF1 in all five cases where it was employed. X-LAG is a new infant-onset gigantism syndrome that has a severe clinical phenotype leading to challenging disease management.
Keywords: FIPA; GPR101; X chromosome; X-LAG syndrome; duplication; gigantism; pediatric; pituitary adenoma.
© 2015 Society for Endocrinology.
Figures
References
-
- Asa SL, Kovacs K, Stefaneanu L, Horvath E, Billestrup N, Gonzalez-Manchon C, Vale W. Pituitary mammosomatotroph adenomas develop in old mice transgenic for growth hormone-releasing hormone. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine. 1990;193:232–235. - PubMed
-
- AvRuskin TW, Sau K, Tang S, Juan C. Childhood acromegaly: successful therapy with conventional radiation and effects of chlorpromazine on growth hormone and prolactin secretion. The Journal of Clinical Endocrinology & Metabolism. 1973;37:380–388. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
