

Interpretation of mRNA splicing mutations in genetic disease: review of the literature and guidelines for information-theoretical analysis
- PMID: 25717368
- PMCID: PMC4329672
- DOI: 10.12688/f1000research.5654.1
Interpretation of mRNA splicing mutations in genetic disease: review of the literature and guidelines for information-theoretical analysis
Abstract
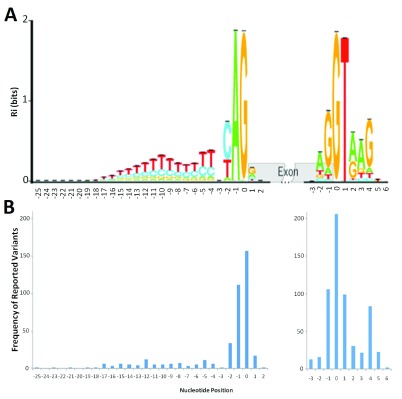
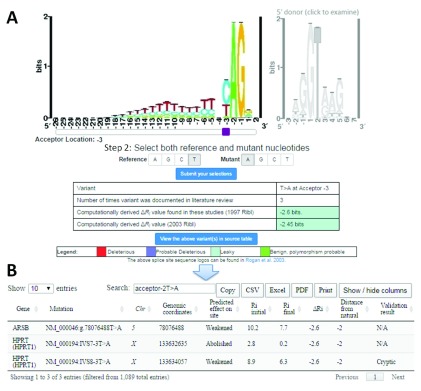
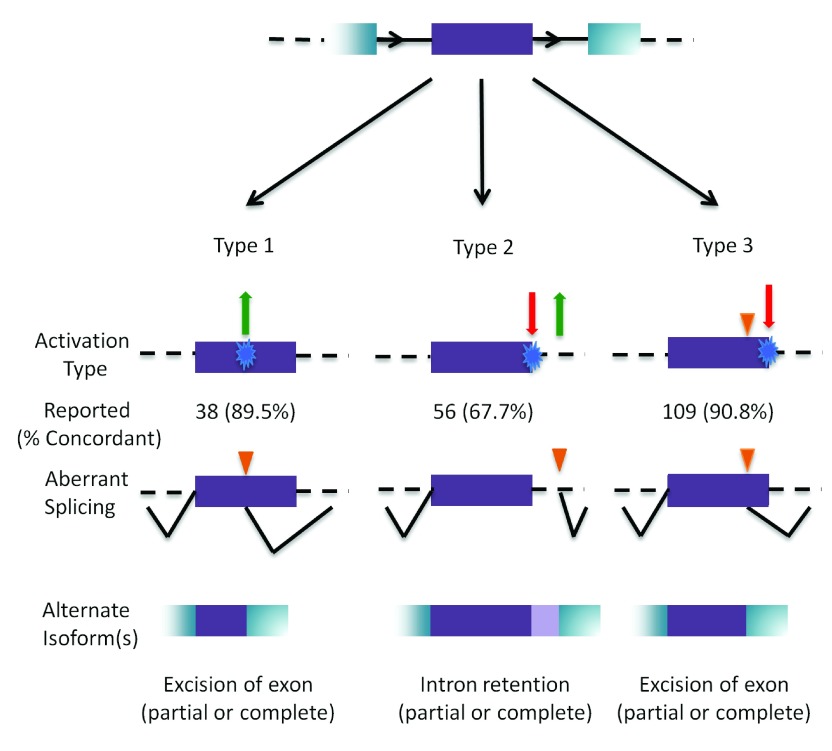
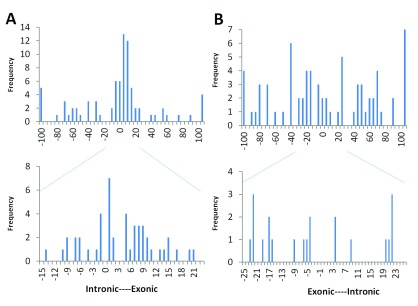
The interpretation of genomic variants has become one of the paramount challenges in the post-genome sequencing era. In this review we summarize nearly 20 years of research on the applications of information theory (IT) to interpret coding and non-coding mutations that alter mRNA splicing in rare and common diseases. We compile and summarize the spectrum of published variants analyzed by IT, to provide a broad perspective of the distribution of deleterious natural and cryptic splice site variants detected, as well as those affecting splicing regulatory sequences. Results for natural splice site mutations can be interrogated dynamically with Splicing Mutation Calculator, a companion software program that computes changes in information content for any splice site substitution, linked to corresponding publications containing these mutations. The accuracy of IT-based analysis was assessed in the context of experimentally validated mutations. Because splice site information quantifies binding affinity, IT-based analyses can discern the differences between variants that account for the observed reduced (leaky) versus abolished mRNA splicing. We extend this principle by comparing predicted mutations in natural, cryptic, and regulatory splice sites with observed deleterious phenotypic and benign effects. Our analysis of 1727 variants revealed a number of general principles useful for ensuring portability of these analyses and accurate input and interpretation of mutations. We offer guidelines for optimal use of IT software for interpretation of mRNA splicing mutations.
Keywords: genetic disease; mRNA; mutation; rare disease; splicing.
Conflict of interest statement
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources