

Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase
- PMID: 25727495
- PMCID: PMC4397983
- DOI: 10.1038/ncomms7250
Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase
Abstract
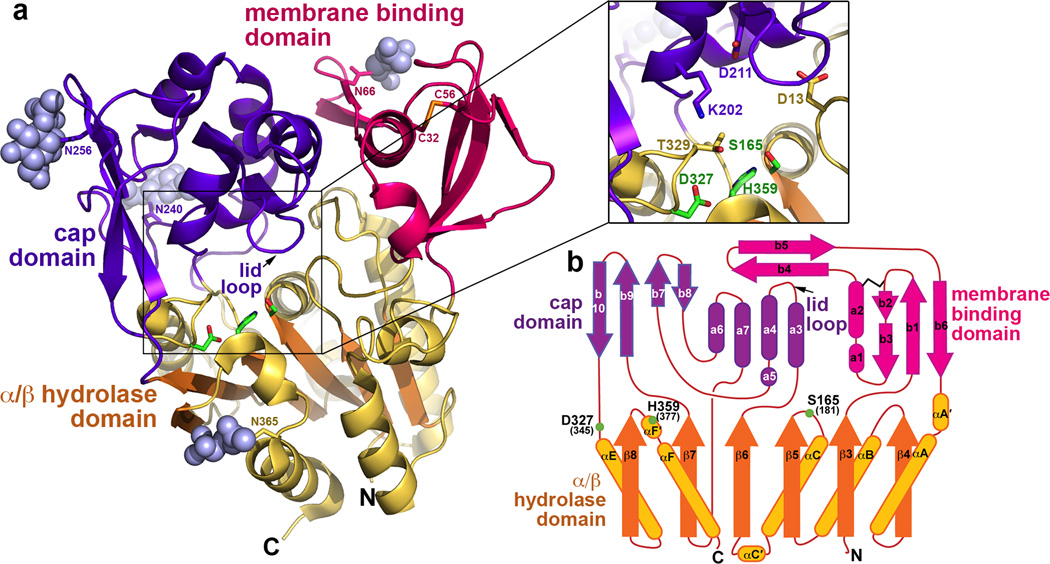
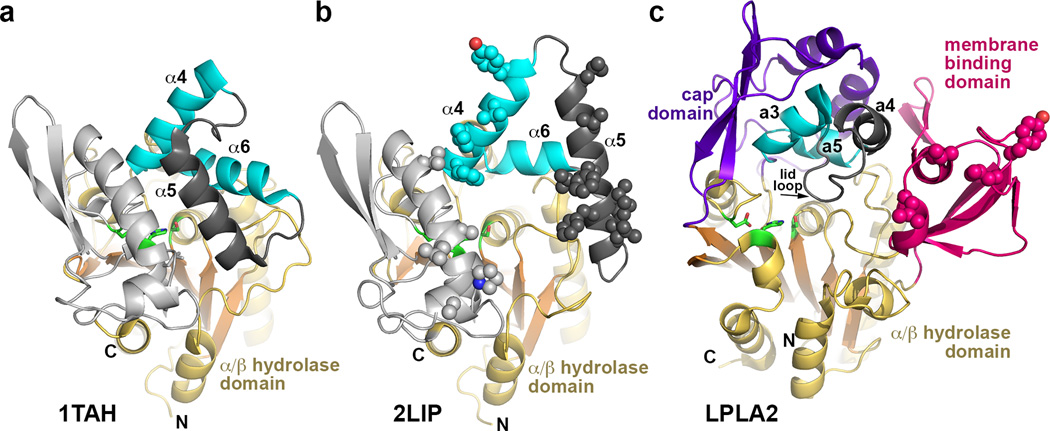
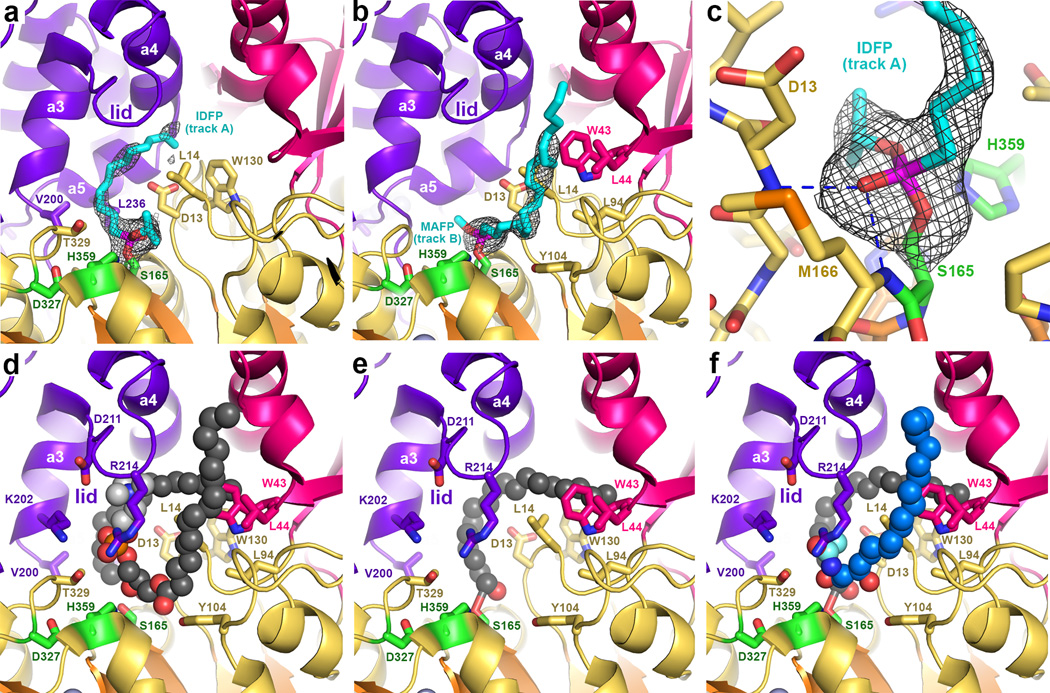
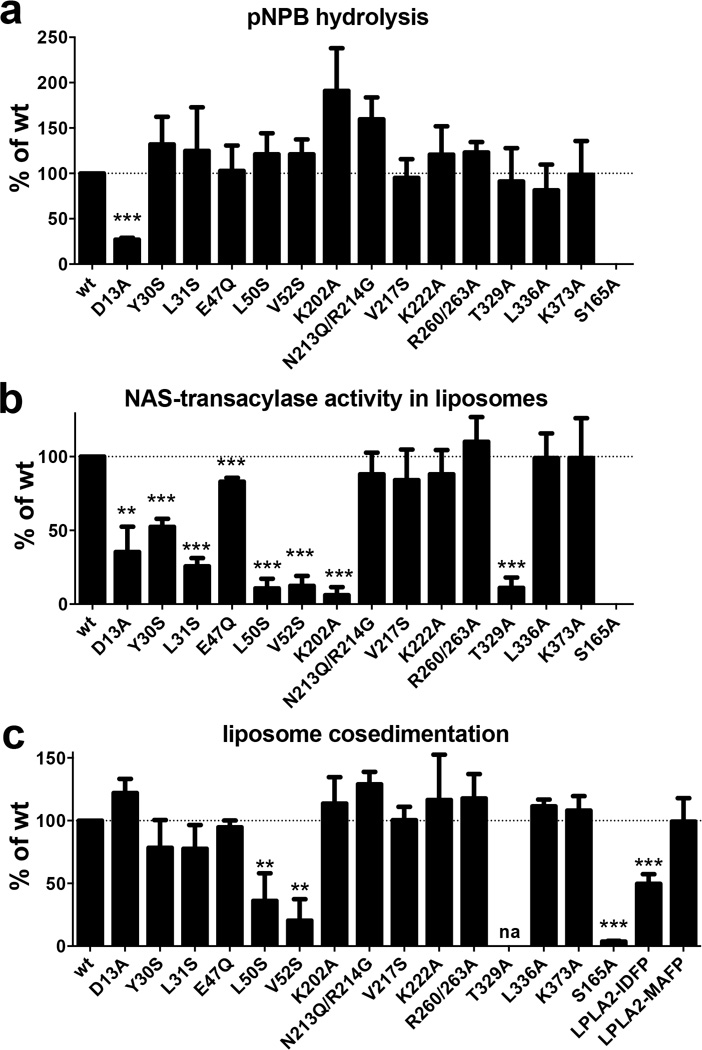
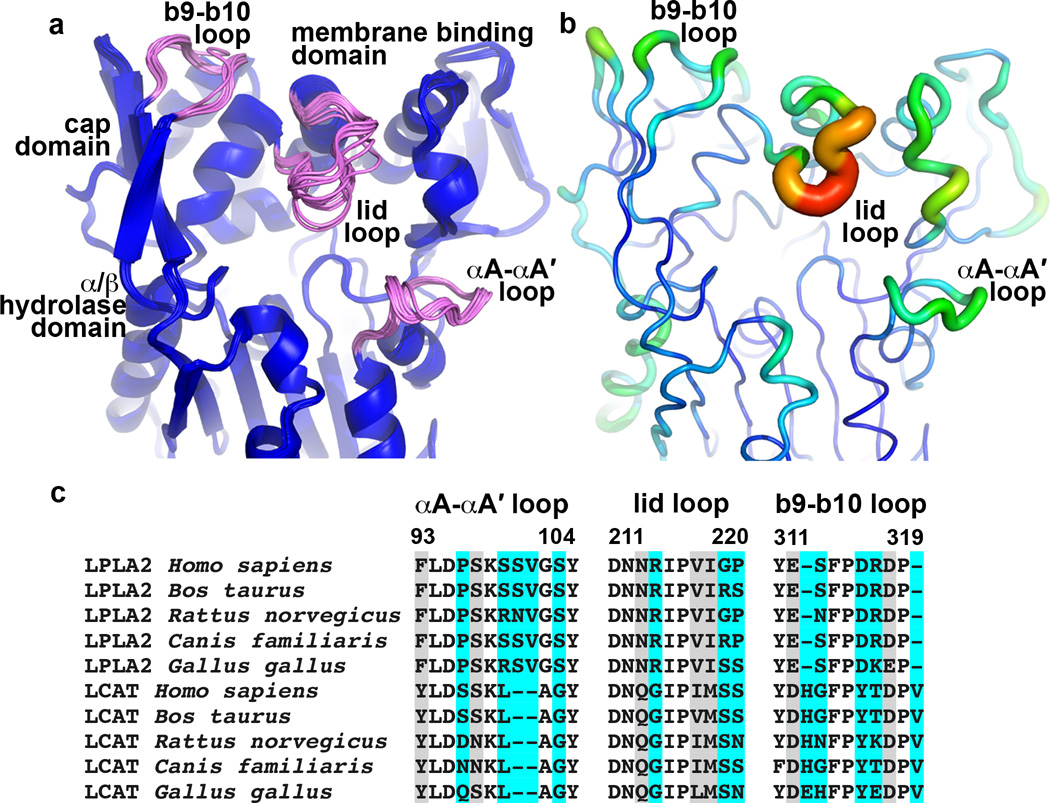
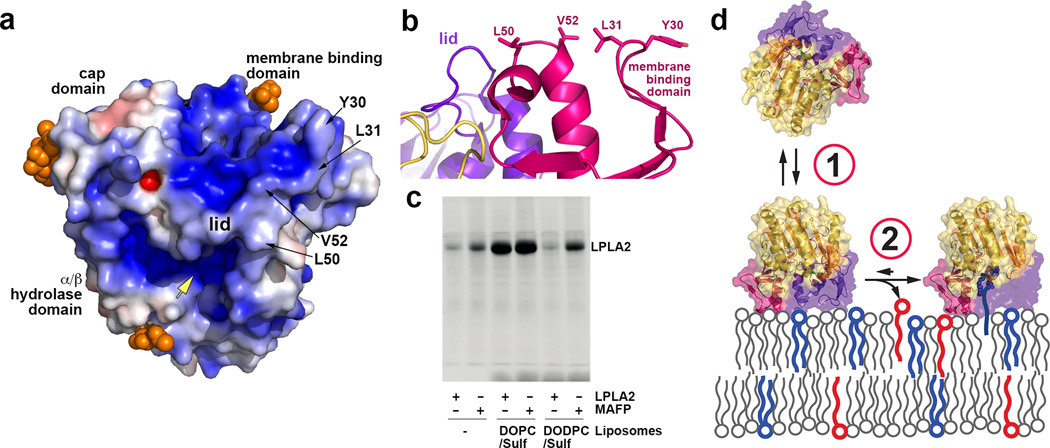
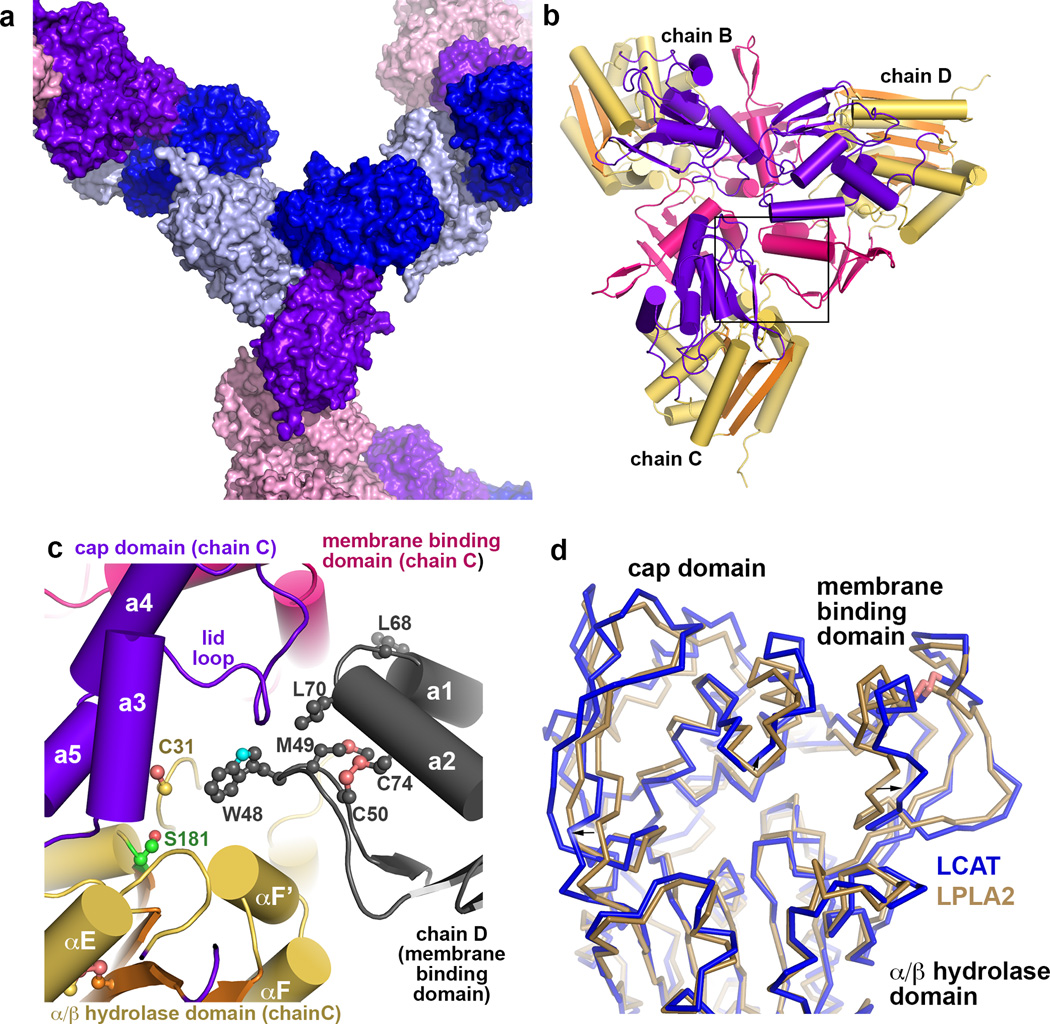
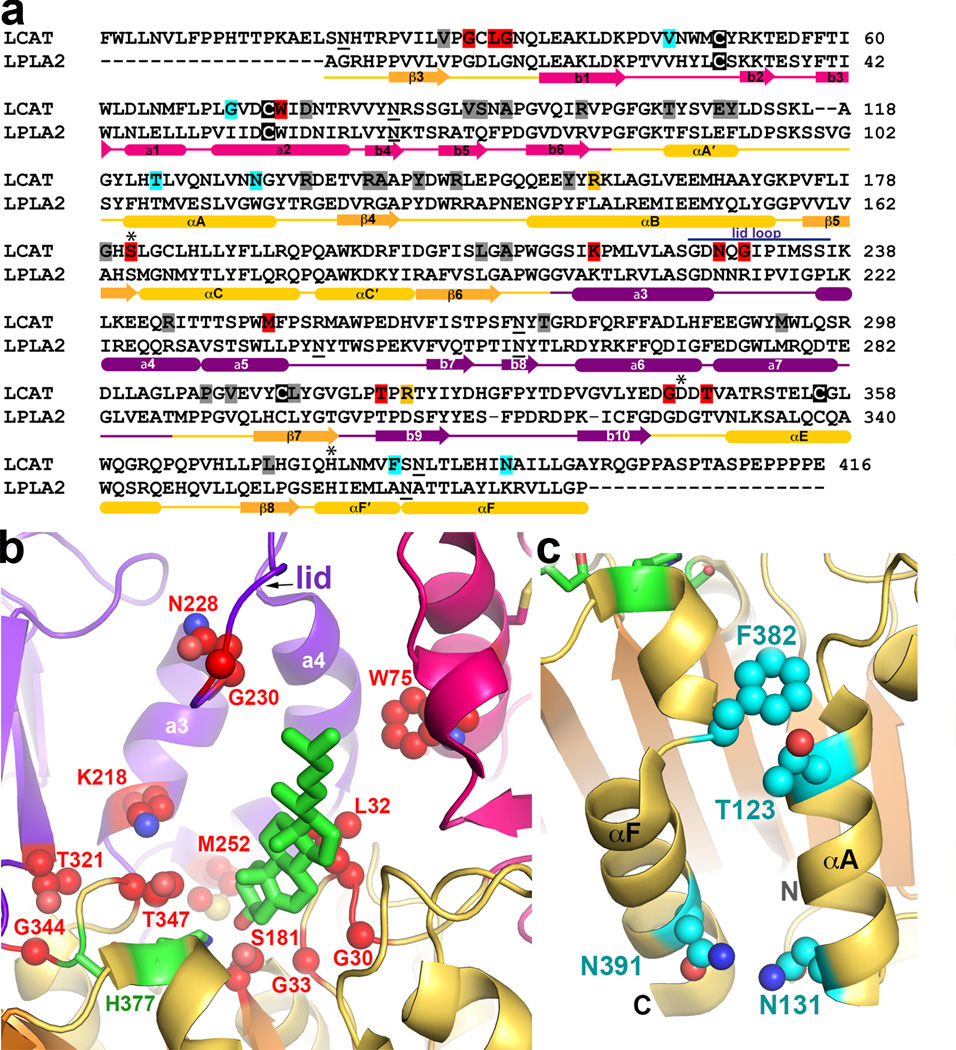
Lysosomal phospholipase A2 (LPLA2) and lecithin:cholesterol acyltransferase (LCAT) belong to a structurally uncharacterized family of key lipid-metabolizing enzymes responsible for lung surfactant catabolism and for reverse cholesterol transport, respectively. Whereas LPLA2 is predicted to underlie the development of drug-induced phospholipidosis, somatic mutations in LCAT cause fish eye disease and familial LCAT deficiency. Here we describe several high-resolution crystal structures of human LPLA2 and a low-resolution structure of LCAT that confirms its close structural relationship to LPLA2. Insertions in the α/β hydrolase core of LPLA2 form domains that are responsible for membrane interaction and binding the acyl chains and head groups of phospholipid substrates. The LCAT structure suggests the molecular basis underlying human disease for most of the known LCAT missense mutations, and paves the way for rational development of new therapeutics to treat LCAT deficiency, atherosclerosis and acute coronary syndrome.
Figures
References
-
- Abe A, et al. Lysosomal phospholipase A2 is selectively expressed in alveolar macrophages. J. Biol. Chem. 2004;279:42605–42611. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
