

Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4
- PMID: 25728773
- PMCID: PMC4375449
- DOI: 10.1016/j.ajhg.2015.01.005
Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4
Abstract
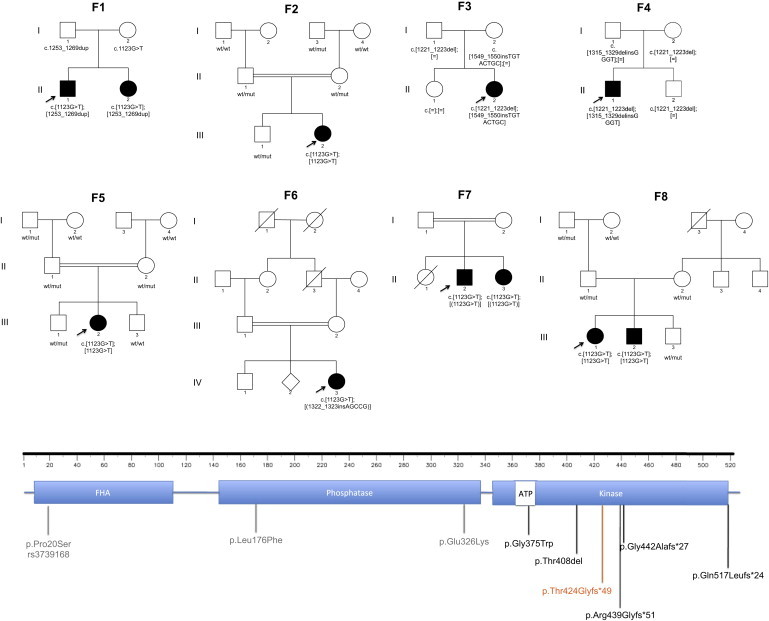
Hereditary autosomal-recessive cerebellar ataxias are a genetically and clinically heterogeneous group of disorders. We used homozygosity mapping and exome sequencing to study a cohort of nine Portuguese families who were identified during a nationwide, population-based, systematic survey as displaying a consistent phenotype of recessive ataxia with oculomotor apraxia (AOA). The integration of data from these analyses led to the identification of the same homozygous PNKP (polynucleotide kinase 3'-phosphatase) mutation, c.1123G>T (p.Gly375Trp), in three of the studied families. When analyzing this particular gene in the exome sequencing data from the remaining cohort, we identified homozygous or compound-heterozygous mutations in five other families. PNKP is a dual-function enzyme with a key role in different pathways of DNA-damage repair. Mutations in this gene have previously been associated with an autosomal-recessive syndrome characterized by microcephaly; early-onset, intractable seizures; and developmental delay (MCSZ). The finding of PNKP mutations associated with recessive AOA extends the phenotype associated with this gene and identifies a fourth locus that causes AOA. These data confirm that MCSZ and some forms of ataxia share etiological features, most likely reflecting the role of PNKP in DNA-repair mechanisms.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Barbot C., Coutinho P., Chorão R., Ferreira C., Barros J., Fineza I., Dias K., Monteiro J., Guimarães A., Mendonça P. Recessive ataxia with ocular apraxia: review of 22 Portuguese patients. Arch. Neurol. 2001;58:201–205. - PubMed
-
- Moreira M.C., Barbot C., Tachi N., Kozuka N., Uchida E., Gibson T., Mendonça P., Costa M., Barros J., Yanagisawa T. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet. 2001;29:189–193. - PubMed
-
- Le Ber I., Bouslam N., Rivaud-Péchoux S., Guimarães J., Benomar A., Chamayou C., Goizet C., Moreira M.C., Klur S., Yahyaoui M. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127:759–767. - PubMed
-
- Moreira M.C., Klur S., Watanabe M., Németh A.H., Le Ber I., Moniz J.C., Tranchant C., Aubourg P., Tazir M., Schöls L. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004;36:225–227. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
