

The Spectrum of C9orf72-mediated Neurodegeneration and Amyotrophic Lateral Sclerosis
- PMID: 25731823
- PMCID: PMC4404438
- DOI: 10.1007/s13311-015-0342-1
The Spectrum of C9orf72-mediated Neurodegeneration and Amyotrophic Lateral Sclerosis
Abstract
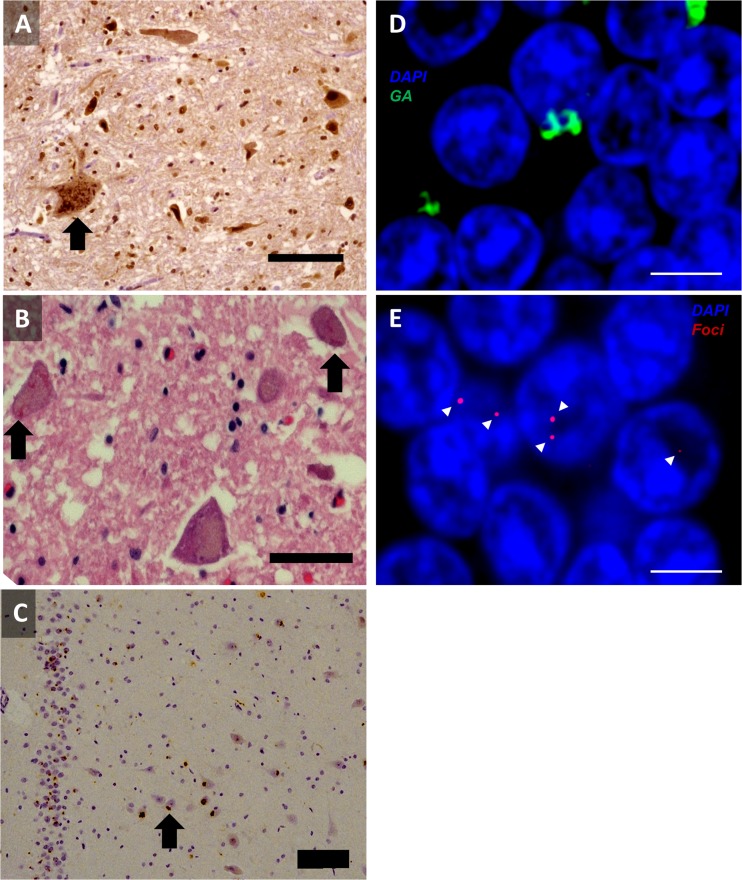
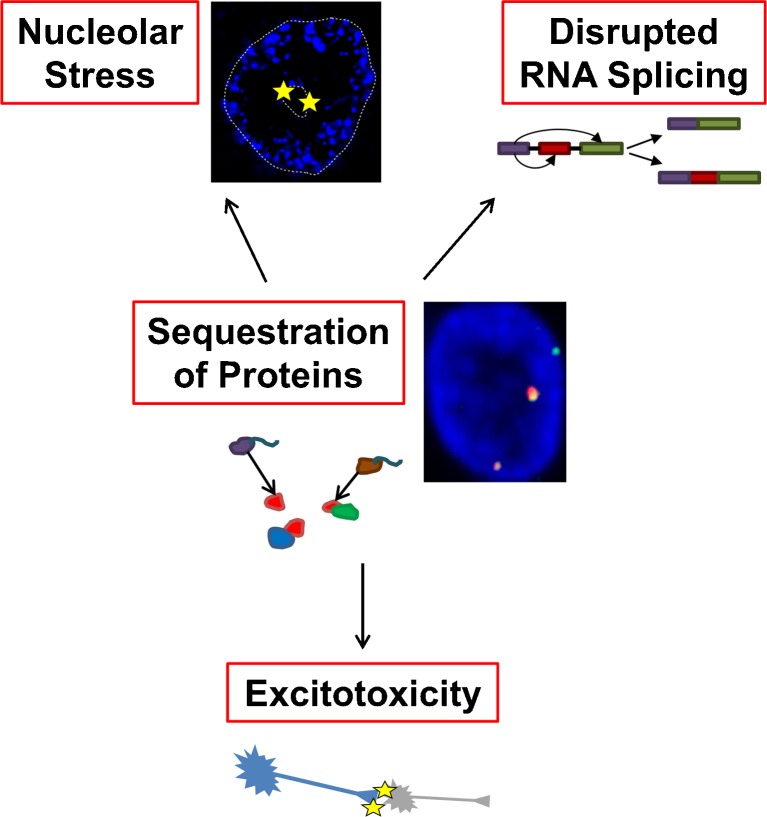
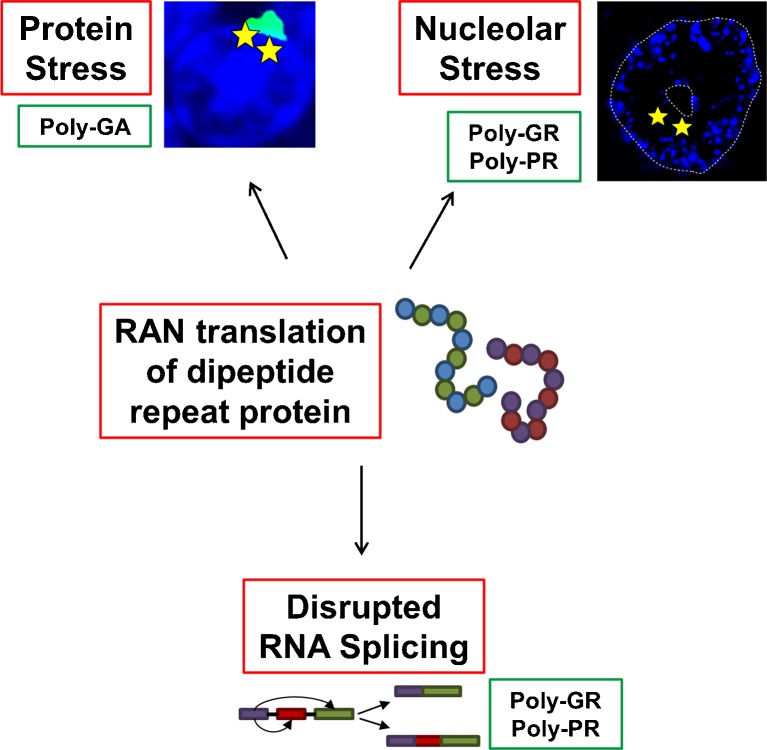
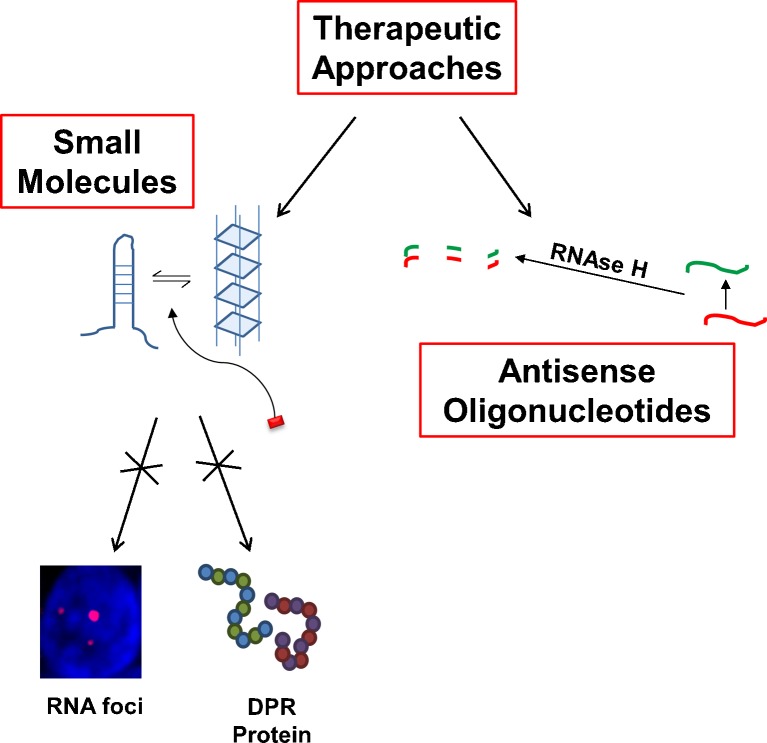
The discovery that a hexanucleotide repeat expansion in C9orf72 is the most numerous genetic variant of both amyotrophic lateral sclerosis and frontotemporal dementia has opened a rapidly growing field, which may provide long hoped for advances in the understanding and treatment of these devastating diseases. In this review we describe the various phenotypes, clinical and pathological, associated with expansion of C9orf72, which go beyond amyotrophic lateral sclerosis and frontotemporal dementia to include neurodegeneration more broadly. Next we take a step back and summarize the current understanding of the C9orf72 expansion and its protein products at a molecular level. Three mechanisms are prominent: toxicity mediated directly by RNA transcribed from the repeat; toxicity mediated by dipeptide repeat proteins translated from the repeat sequence; and haploinsufficiency resulting from reduced transcription of the C9orf72 exonic sequence. A series of exciting advances have recently described how dipeptide repeat proteins might interfere with the normal role of the nucleolus in maturation of RNA binding proteins and in production of ribosomes. Importantly, these mechanisms are unlikely to be mutually exclusive. We draw attention to the fact that clinical and pathological similarities to other genetic variants without a repeat expansion must not be overlooked in ascribing a pathogenic mechanism to C9orf72-disease. Finally, with a view to impact on patient care, we discuss current practice with respect to genetic screening in patients with and without a family history of disease, and the most promising developments towards therapy that have been reported to date.
Figures
References
-
- Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6:37–46. - PubMed
-
- Gijselinck I, Engelborghs S, Maes G, et al. Identification of 2 loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:606–616. - PubMed
-
- Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
