

Structure and function of the bacterial root microbiota in wild and domesticated barley
- PMID: 25732064
- PMCID: PMC4362959
- DOI: 10.1016/j.chom.2015.01.011
Structure and function of the bacterial root microbiota in wild and domesticated barley
Abstract
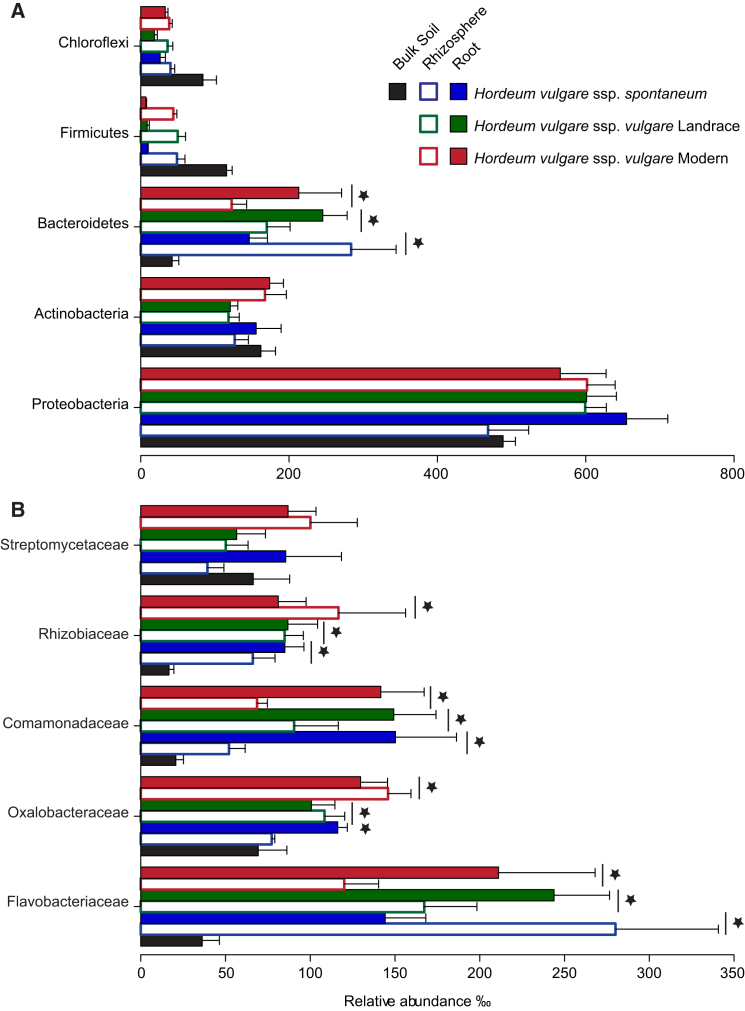
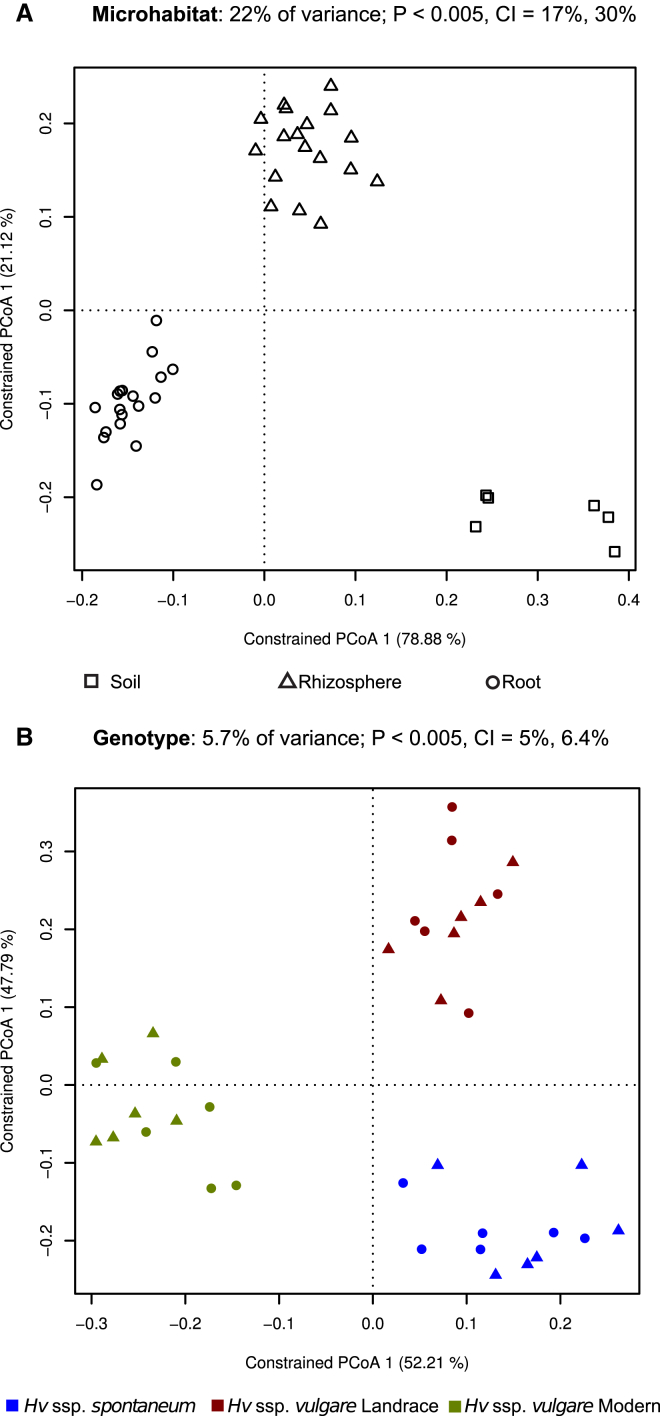
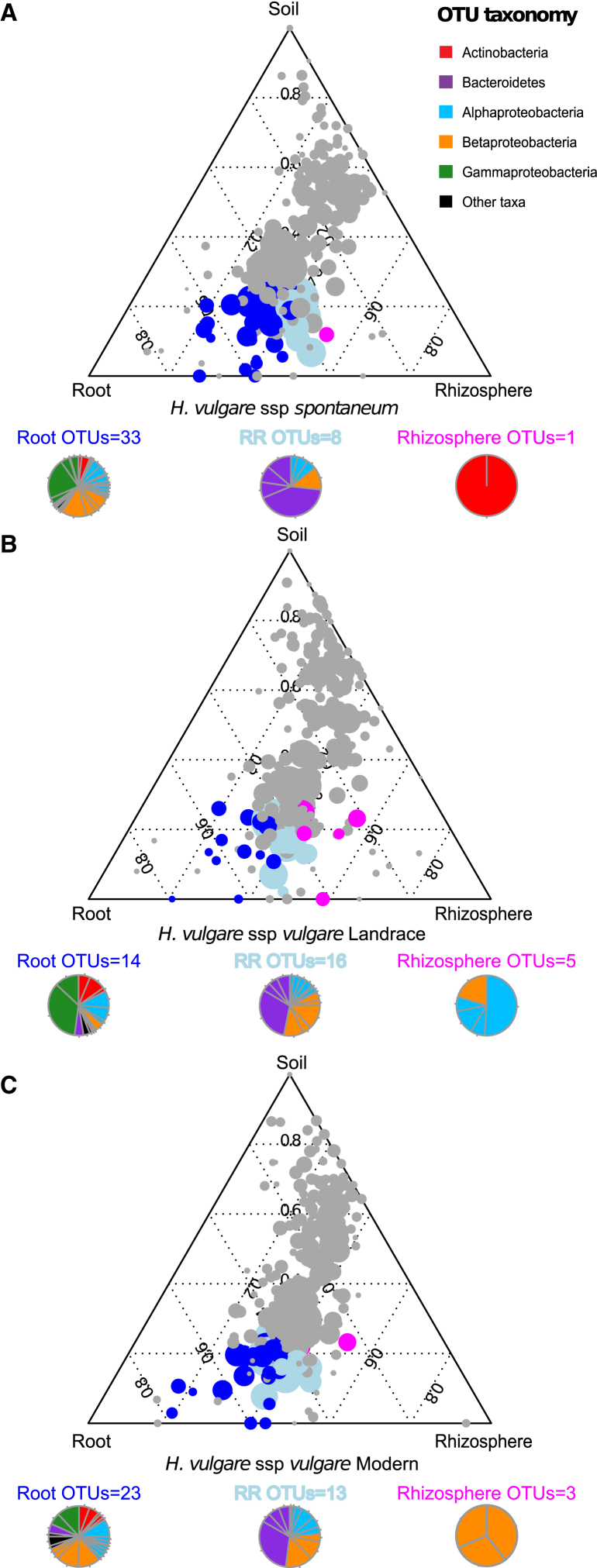
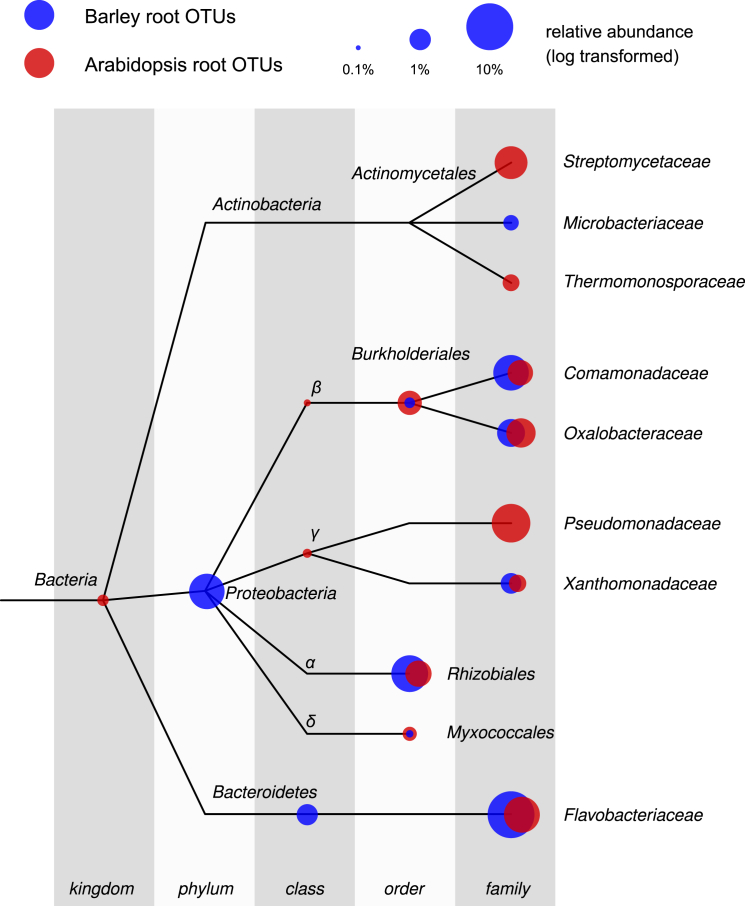
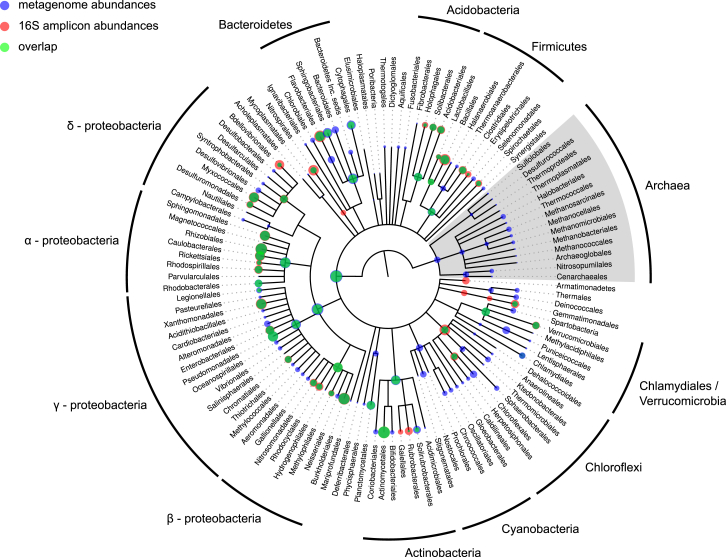
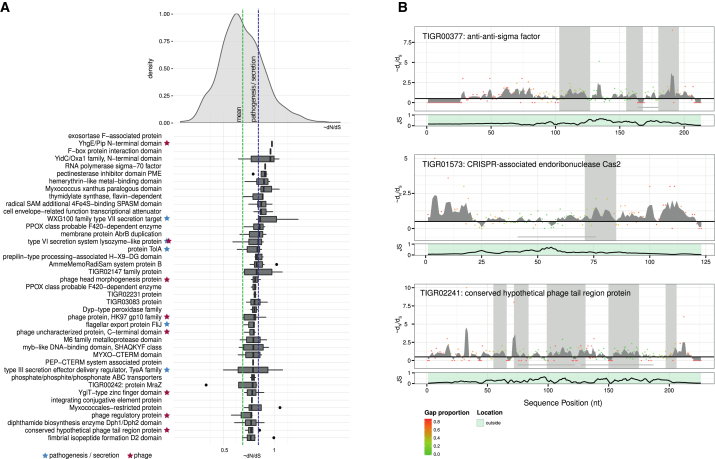
The microbial communities inhabiting the root interior of healthy plants, as well as the rhizosphere, which consists of soil particles firmly attached to roots, engage in symbiotic associations with their host. To investigate the structural and functional diversification among these communities, we employed a combination of 16S rRNA gene profiling and shotgun metagenome analysis of the microbiota associated with wild and domesticated accessions of barley (Hordeum vulgare). Bacterial families Comamonadaceae, Flavobacteriaceae, and Rhizobiaceae dominate the barley root-enriched microbiota. Host genotype has a small, but significant, effect on the diversity of root-associated bacterial communities, possibly representing a footprint of barley domestication. Traits related to pathogenesis, secretion, phage interactions, and nutrient mobilization are enriched in the barley root-associated microbiota. Strikingly, protein families assigned to these same traits showed evidence of positive selection. Our results indicate that the combined action of microbe-microbe and host-microbe interactions drives microbiota differentiation at the root-soil interface.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Abbo S., Pinhasi van-Oss R., Gopher A., Saranga Y., Ofner I., Peleg Z. Plant domestication versus crop evolution: a conceptual framework for cereals and grain legumes. Trends Plant Sci. 2014;19:351–360. - PubMed
-
- Anderson M.J., Willis T.J. Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology. 2003;84:511–525.
-
- Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. - PubMed
-
- Boller T., Felix G. A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 2009;60:379–406. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
