

Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics
- PMID: 25732828
- PMCID: PMC4542311
- DOI: 10.1016/j.celrep.2015.02.001
Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics
Abstract
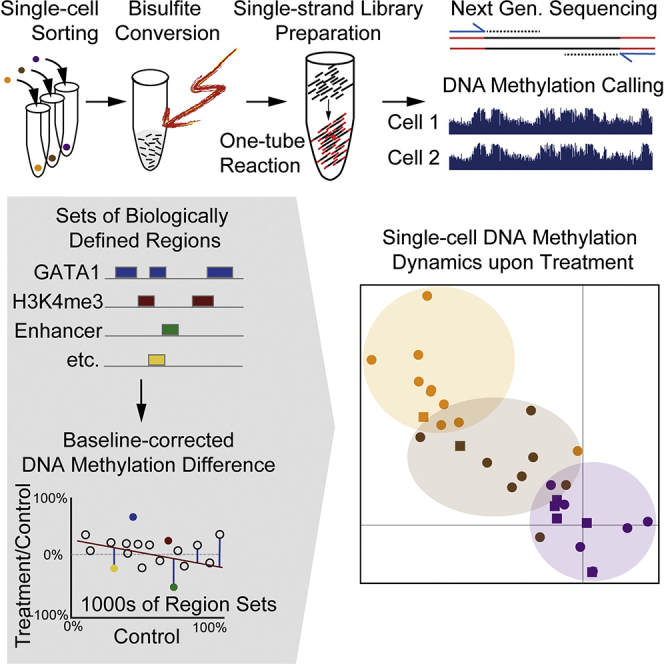
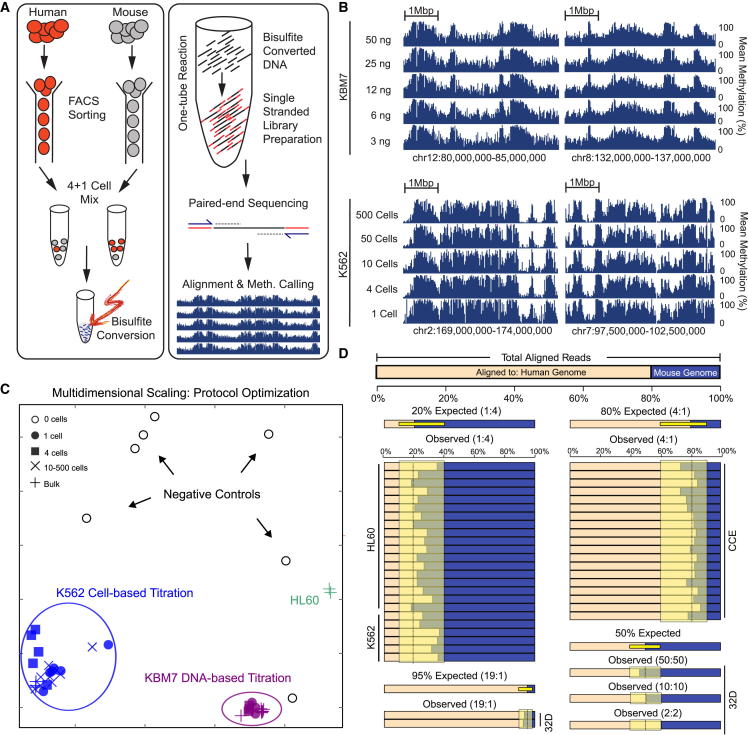
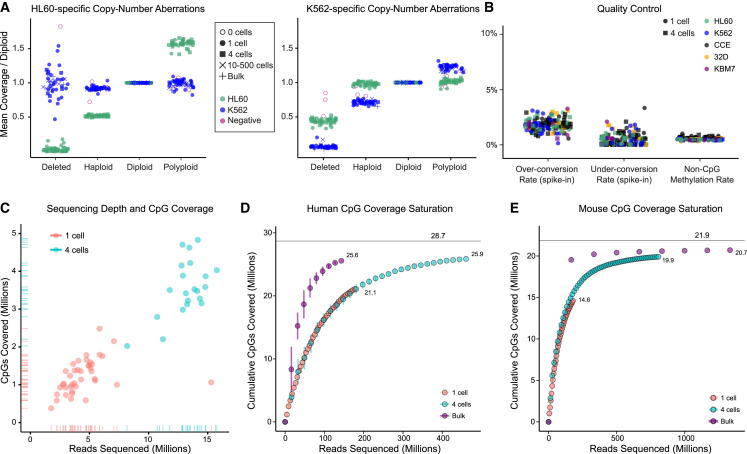
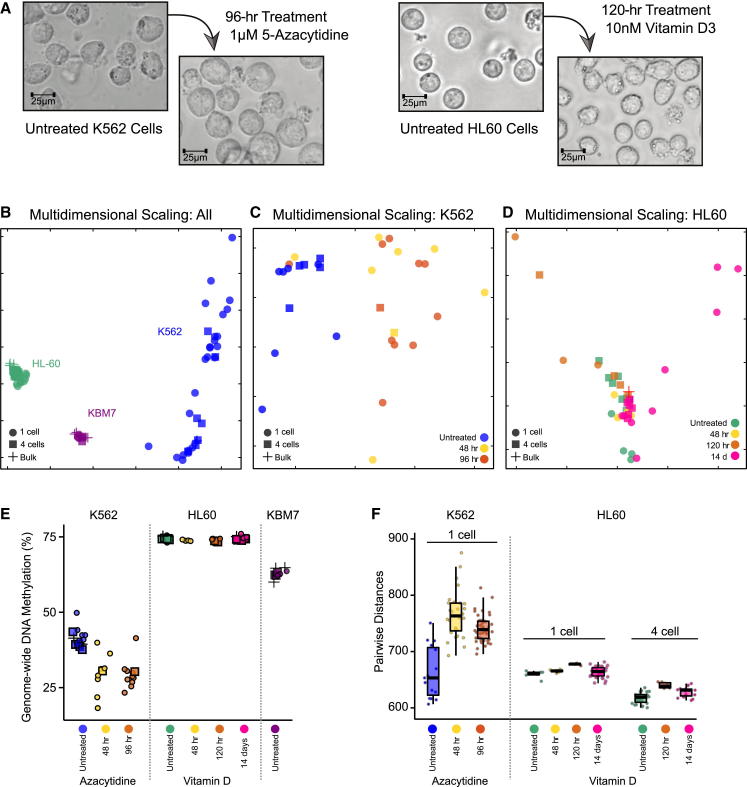
Methods for single-cell genome and transcriptome sequencing have contributed to our understanding of cellular heterogeneity, whereas methods for single-cell epigenomics are much less established. Here, we describe a whole-genome bisulfite sequencing (WGBS) assay that enables DNA methylation mapping in very small cell populations (μWGBS) and single cells (scWGBS). Our assay is optimized for profiling many samples at low coverage, and we describe a bioinformatic method that analyzes collections of single-cell methylomes to infer cell-state dynamics. Using these technological advances, we studied epigenomic cell-state dynamics in three in vitro models of cellular differentiation and pluripotency, where we observed characteristic patterns of epigenome remodeling and cell-to-cell heterogeneity. The described method enables single-cell analysis of DNA methylation in a broad range of biological systems, including embryonic development, stem cell differentiation, and cancer. It can also be used to establish composite methylomes that account for cell-to-cell heterogeneity in complex tissue samples.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
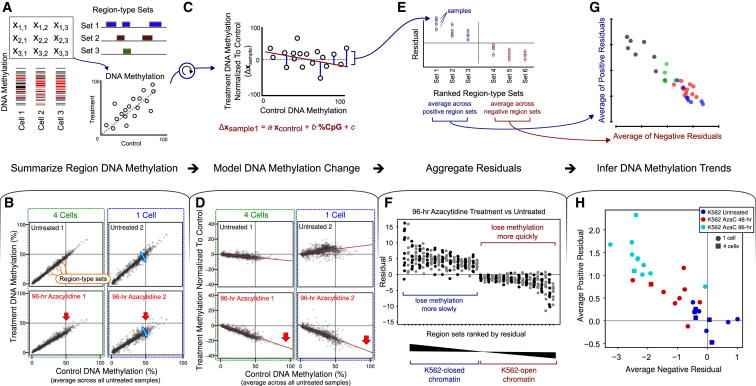
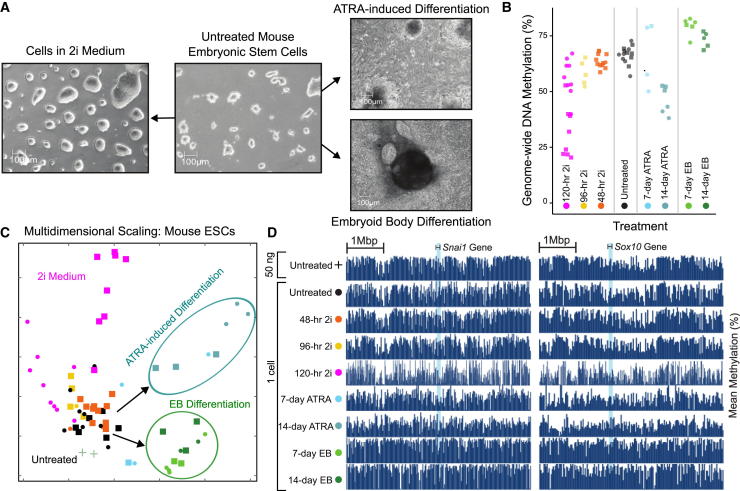
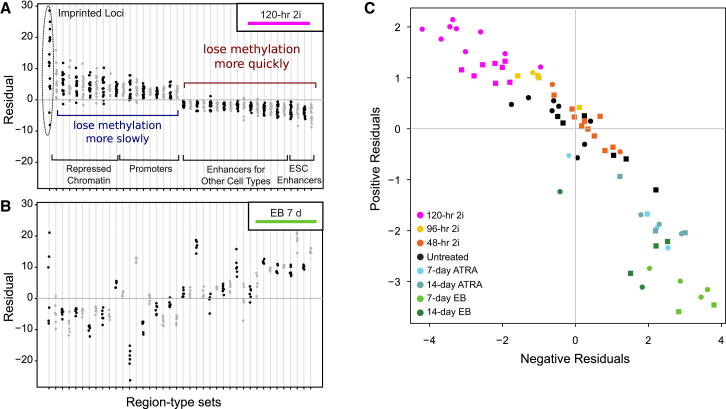
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
