

Review
doi: 10.1083/jcb.201409063.
The cell biology of fat expansion
Affiliations
- PMID: 25733711
- PMCID: PMC4347644
- DOI: 10.1083/jcb.201409063
Item in Clipboard
Review
The cell biology of fat expansion
J Cell Biol.
.
Abstract
Adipose tissue is a complex, multicellular organ that profoundly influences the function of nearly all other organ systems through its diverse metabolite and adipokine secretome. Adipocytes are the primary cell type of adipose tissue and play a key role in maintaining energy homeostasis. The efficiency with which adipose tissue responds to whole-body energetic demands reflects the ability of adipocytes to adapt to an altered nutrient environment, and has profound systemic implications. Deciphering adipocyte cell biology is an important component of understanding how the aberrant physiology of expanding adipose tissue contributes to the metabolic dysregulation associated with obesity.
© 2015 Rutkowski et al.
Figures
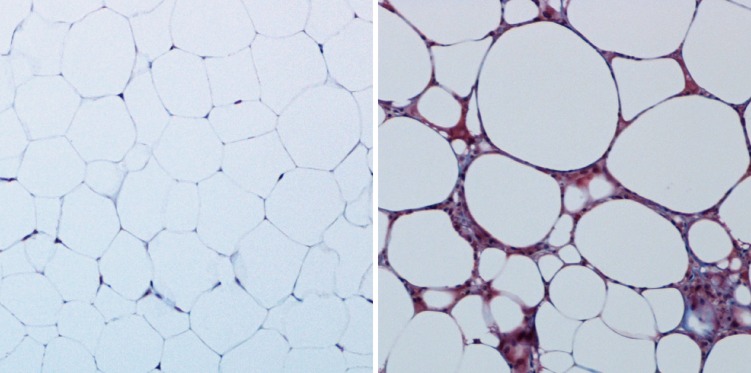
Benign and unhealthy adipose tissue. The overall health of adipose tissue can be visualized histologically. Benign murine subcutaneous adipose tissue (left; trichrome staining) is rich in unilocular white adipocytes with sparse ECM. On high-fat feeding (right), adipose tissue experiences hypoxia and inflammation, resulting in a dense, fibrous ECM. These histological tissue features are characteristic of insulin-resistant adipocytes.
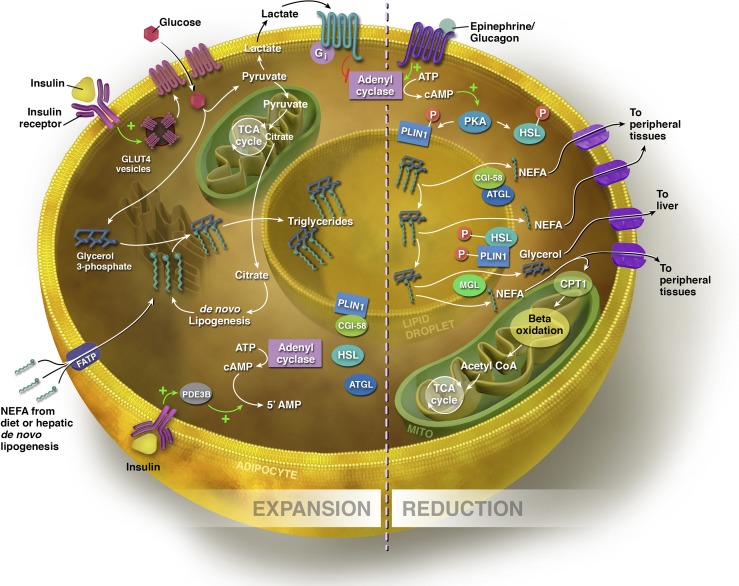
Expansion and reduction of the adipocyte. Sources of fatty acids for triglyceride synthesis within the adipocyte include exogenous NEFAs from the diet and those synthesized via de novo lipogenesis in the hepatocyte and adipocyte. NEFAs can enter the adipocyte via FATPs. In the presence of glucose, insulin binds to its receptor, stimulating the translocation of GLUT-4 transporters from the cytosol to the cell membrane. Increased flux through glycolysis leads to excess citrate generated in the TCA cycle, which is diverted into the cytosol for fatty acid synthesis. To synthesize triglyceride, three NEFAs are esterified to a glycerol backbone, and the triglyceride is processed through the ER for storage within the lipid droplet. In the expansive state, lipolysis is inhibited by insulin. PLIN1 is bound to CGI-58, the co-activator of ATGL, the rate-limiting enzyme of lipolysis. Through insulin-stimulated activation of phosphodiesterase 3B (PDE3B), which controls intracellular cAMP pools by converting cAMP to the inactive 5′ AMP, insulin prevents the phosphorylation of ATGL and HSL. Insulin also acts on the adipocyte via glucose flux to lactate to inhibit lipolysis indirectly via a lactate/GPR81-dependent inhibition of cAMP. In the lipolytic state, triglycerides are hydrolyzed to release NEFAs, and the size of the lipid droplet is reduced. Via β-AR signaling, Plin1 and HSL are phosphorylated by PKA, stimulating the release of CGI-58. ATGL moves to the lipid droplet surface to hydrolyze the first fatty acid from the triglyceride. Phosphorylated HSL binds Plin1 and hydrolyzes the resulting diacylglycerol to a monoacylglycerol, which is subsequently hydrolyzed by monoacylglycerol lipase (MGL), producing a third fatty acid and glycerol molecule. NEFAs are either exported to other tissues to undergo β-oxidation or re-esterification, or undergo β-oxidation within the mitochondrial matrix of adipocyte. Glycerol is transported to the liver, where it can be converted to glycolytic intermediates or phosphorylated to G3P for triglyceride synthesis (see text for details).
References
-
- Agarwal A.K., Sukumaran S., Cortés V.A., Tunison K., Mizrachi D., Sankella S., Gerard R.D., Horton J.D., and Garg A.. 2011. Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2-/- gene lipodystrophic mice. J. Biol. Chem. 286:37676–37691 10.1074/jbc.M111.250449 - DOI - PMC - PubMed
-
- Alligier M., Meugnier E., Debard C., Lambert-Porcheron S., Chanseaume E., Sothier M., Loizon E., Hssain A.A., Brozek J., Scoazec J.Y., et al. 2012. Subcutaneous adipose tissue remodeling during the initial phase of weight gain induced by overfeeding in humans. J. Clin. Endocrinol. Metab. 97:E183–E192 10.1210/jc.2011-2314 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
