

The yin and yang of kidney development and Wilms' tumors
- PMID: 25737276
- PMCID: PMC4358399
- DOI: 10.1101/gad.256396.114
The yin and yang of kidney development and Wilms' tumors
Abstract
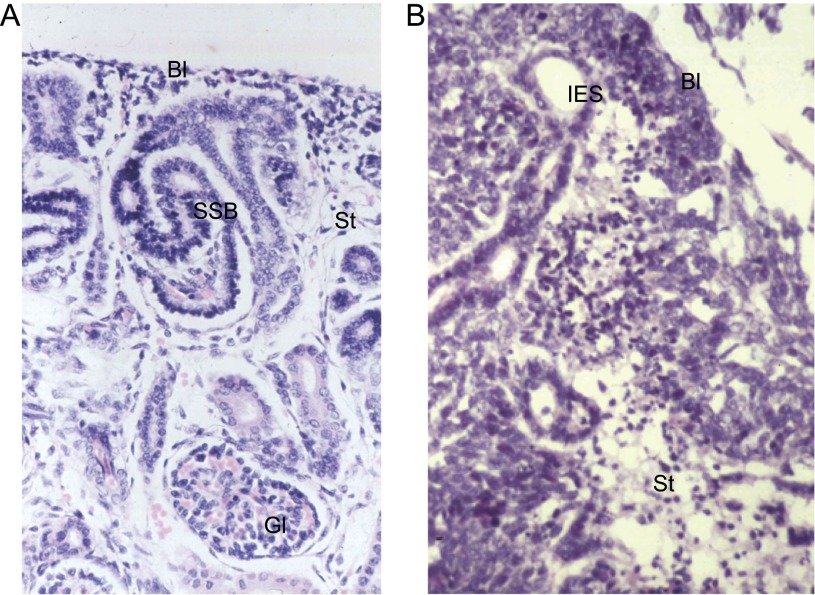
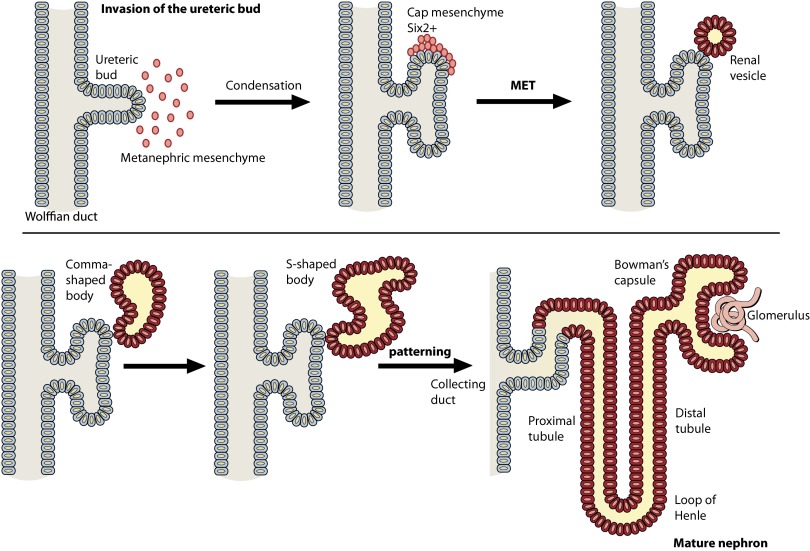
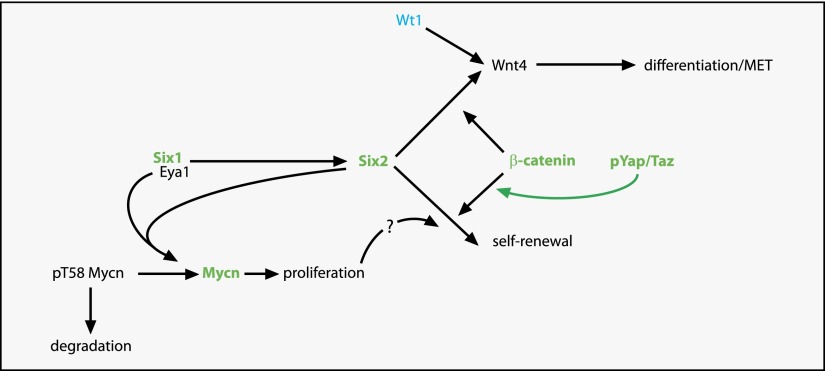
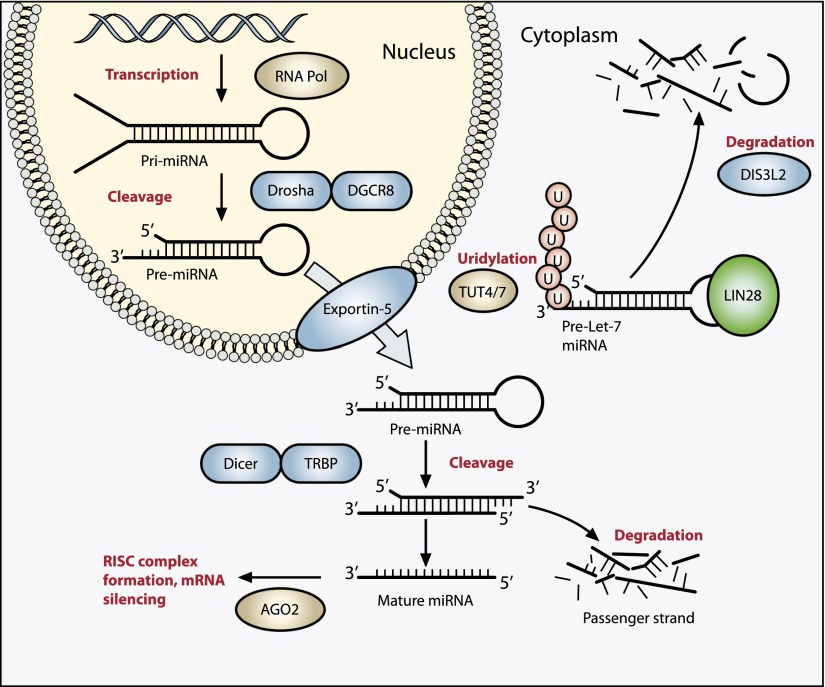
Wilms' tumor, or nephroblastoma, is the most common pediatric renal cancer. The tumors morphologically resemble embryonic kidneys with a disrupted architecture and are associated with undifferentiated metanephric precursors. Here, we discuss genetic and epigenetic findings in Wilms' tumor in the context of renal development. Many of the genes implicated in Wilms' tumorigenesis are involved in the control of nephron progenitors or the microRNA (miRNA) processing pathway. Whereas the first group of genes has been extensively studied in normal development, the second finding suggests important roles for miRNAs in general-and specific miRNAs in particular-in normal kidney development that still await further analysis. The recent identification of Wilms' tumor cancer stem cells could provide a framework to integrate these pathways and translate them into new or improved therapeutic interventions.
Keywords: Wilms’ tumor; Wt1; kidney development; miRNA; nephron progenitor cells; β-catenin.
© 2015 Hohenstein et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Anglesio MS, Evdokimova V, Melnyk N, Zhang L, Fernandez CV, Grundy PE, Leach S, Marra MA, Brooks-Wilson AR, Penninger J, et al. . 2004. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum Mol Genet 13: 2061–2074. - PubMed
-
- Astuti D, Morris MR, Cooper WN, Staals RH, Wake NC, Fews GA, Gill H, Gentle D, Shuib S, Ricketts CJ, et al. . 2012. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44: 277–284. - PubMed
-
- Bahubeshi A, Bal N, Rio Frio T, Hamel N, Pouchet C, Yilmaz A, Bouron-Dal Soglio D, Williams GM, Tischkowitz M, Priest JR, et al. . 2010. Germline DICER1 mutations and familial cystic nephroma. J Med Genet 47: 863–866. - PubMed
-
- Bardeesy N, Falkoff D, Petruzzi M-J, Nowak N, Zabel B, Adam M, Aguiar MC, Grundy P, Shows T, Pelletier J. 1994. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 7: 91–97. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical