

Mitochondrial dynamics in diabetic cardiomyopathy
- PMID: 25738230
- PMCID: PMC4449634
- DOI: 10.1089/ars.2015.6293
Mitochondrial dynamics in diabetic cardiomyopathy
Abstract
Significance: Cardiac function is energetically demanding, reliant on efficient well-coupled mitochondria to generate adenosine triphosphate and fulfill the cardiac demand. Predictably then, mitochondrial dysfunction is associated with cardiac pathologies, often related to metabolic disease, most commonly diabetes. Diabetic cardiomyopathy (DCM), characterized by decreased left ventricular function, arises independently of coronary artery disease and atherosclerosis. Dysregulation of Ca(2+) handling, metabolic changes, and oxidative stress are observed in DCM, abnormalities reflected in alterations in mitochondrial energetics. Cardiac tissue from DCM patients also presents with altered mitochondrial morphology, suggesting a possible role of mitochondrial dynamics in its pathological progression.
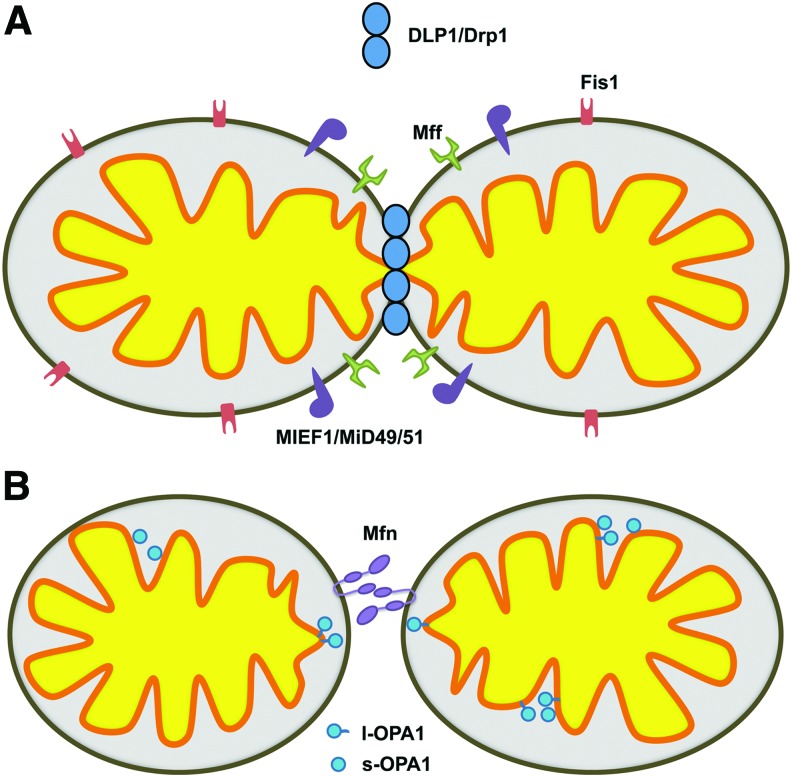
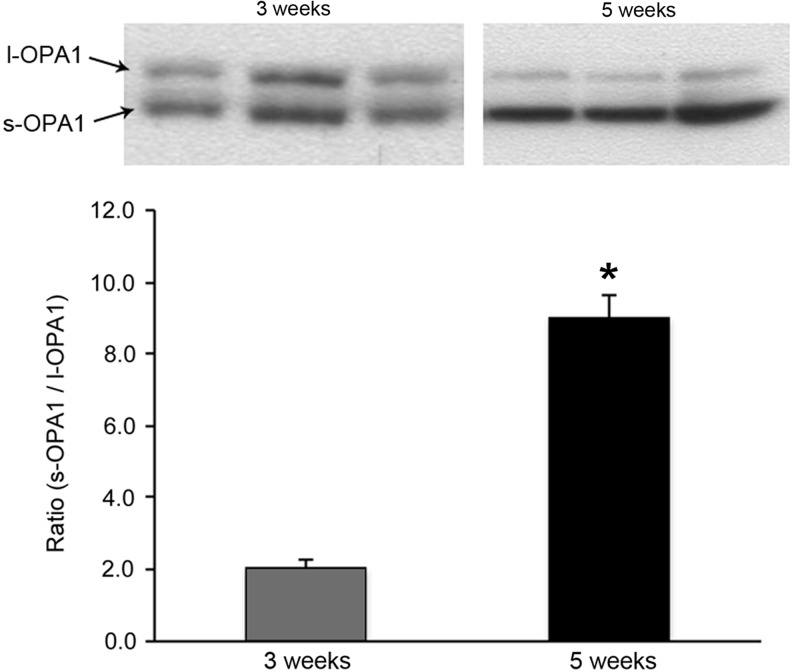
Recent advances: Abnormal mitochondrial morphology is associated with pathologies across diverse tissues, suggesting that this highly regulated process is essential for proper cell maintenance and physiological homeostasis. Highly structured cardiac myofibers were hypothesized to limit alterations in mitochondrial morphology; however, recent work has identified morphological changes in cardiac tissue, specifically in DCM.
Critical issues: Mitochondrial dysfunction has been reported independently from observations of altered mitochondrial morphology in DCM. The temporal relationship and causative nature between functional and morphological changes of mitochondria in the establishment/progression of DCM is unclear.
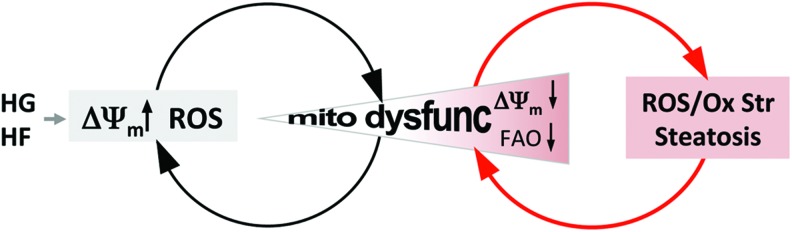
Future directions: Altered mitochondrial energetics and morphology are not only causal for but also consequential to reactive oxygen species production, hence exacerbating oxidative damage through reciprocal amplification, which is integral to the progression of DCM. Therefore, targeting mitochondria for DCM will require better mechanistic characterization of morphological distortion and bioenergetic dysfunction.
Figures




References
-
- Aon MA, Cortassa S, Marban E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278: 44735–44744, 2003 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
