

The direct arylation of allylic sp(3) C-H bonds via organic and photoredox catalysis
- PMID: 25739630
- PMCID: PMC4378681
- DOI: 10.1038/nature14255
The direct arylation of allylic sp(3) C-H bonds via organic and photoredox catalysis
Abstract
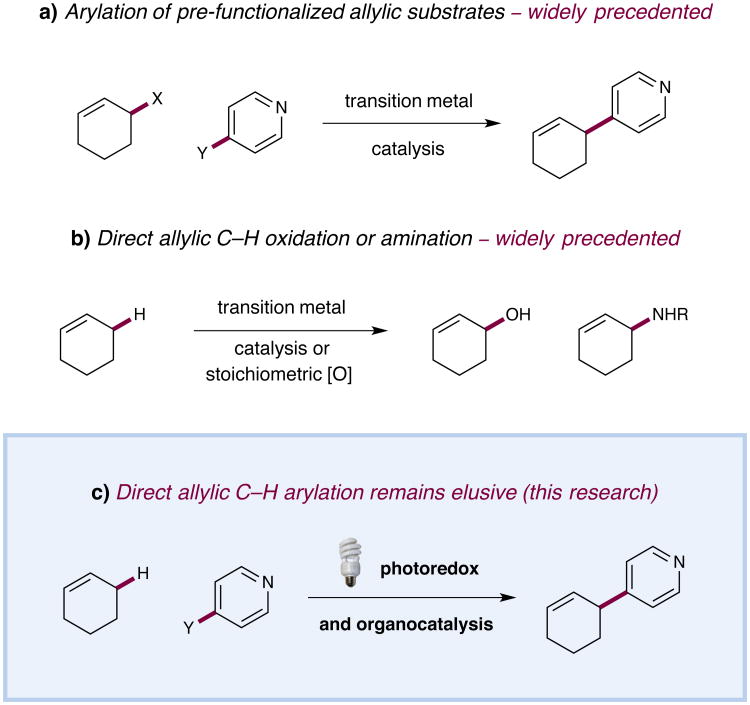
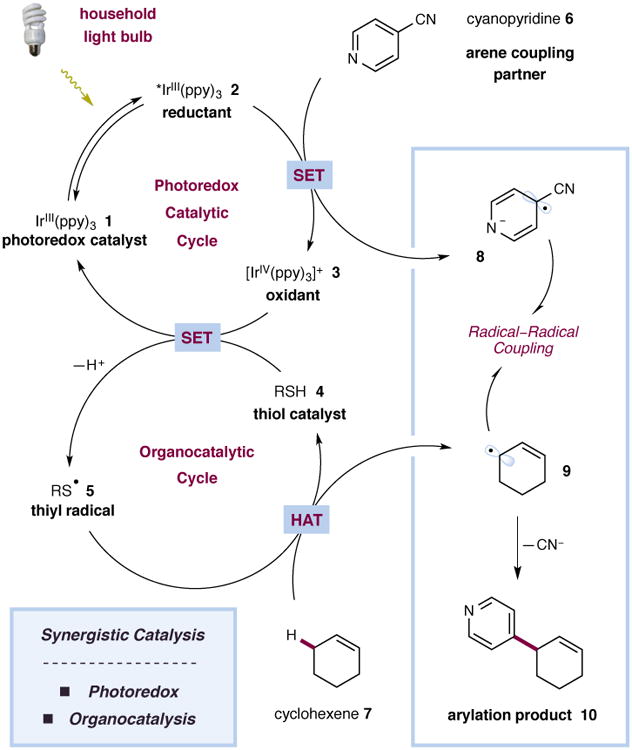
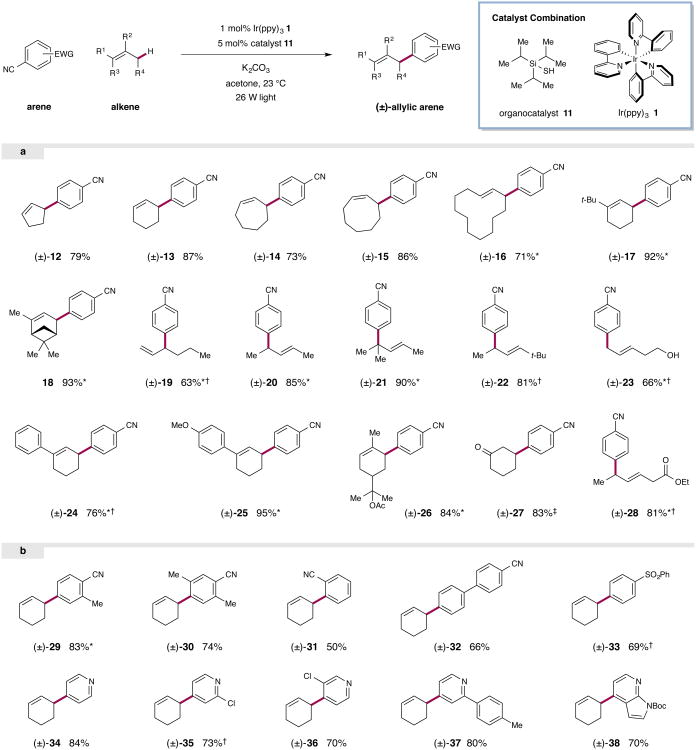
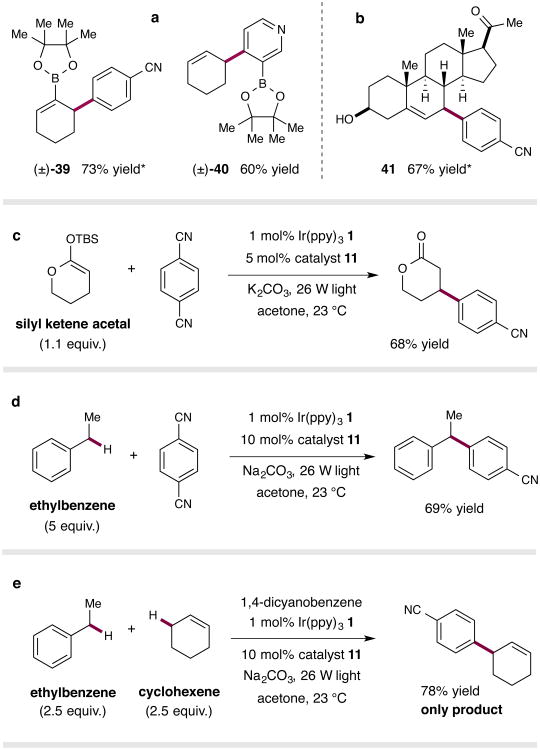
The direct functionalization of unactivated sp(3) C-H bonds is still one of the most challenging problems facing synthetic organic chemists. The appeal of such transformations derives from their capacity to facilitate the construction of complex organic molecules via the coupling of simple and otherwise inert building blocks, without introducing extraneous functional groups. Despite notable recent efforts, the establishment of general and mild strategies for the engagement of sp(3) C-H bonds in C-C bond forming reactions has proved difficult. Within this context, the discovery of chemical transformations that are able to directly functionalize allylic methyl, methylene and methine carbons in a catalytic manner is a priority. Although protocols for direct oxidation and amination of allylic C-H bonds (that is, C-H bonds where an adjacent carbon is involved in a C = C bond) have become widely established, the engagement of allylic substrates in C-C bond forming reactions has thus far required the use of pre-functionalized coupling partners. In particular, the direct arylation of non-functionalized allylic systems would enable access to a series of known pharmacophores (molecular features responsible for a drug's action), though a general solution to this long-standing challenge remains elusive. Here we report the use of both photoredox and organic catalysis to accomplish a mild, broadly effective direct allylic C-H arylation. This C-C bond forming reaction readily accommodates a broad range of alkene and electron-deficient arene reactants, and has been used in the direct arylation of benzylic C-H bonds.
Figures
Comment in
-
Catalysis: dual catalysis at the flick of a switch.Nature. 2015 Mar 5;519(7541):42-3. doi: 10.1038/519042a. Nature. 2015. PMID: 25739627 No abstract available.
References
-
- Eames J, Watkinson M. Catalytic allylic oxidation of alkenes using an asymmetric Kharasch-Sosnovsky reaction. Angew Chem Int Ed. 2001;40:3567–3571. - PubMed
-
- Johannsen M, Jørgensen KA. Allylic Amination. Chem Rev. 1998;98:1689–1708. - PubMed
-
- Trost BM, Van Vranken DL. Asymmetric transition metal-catalyzed allylic alkylations. Chem Rev. 1996;96:395–422. - PubMed
-
- Borg RM, Arnold DR, Cameron TS. Radical ions in photochemistry. 15. The photosubstitution reaction between dicyanobenzenes and alkyl olefins. Can J Chem. 1984;62:1785–1802.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
