

De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose
- PMID: 25740151
- PMCID: PMC4832395
- DOI: 10.1111/brv.12178
De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose
Abstract
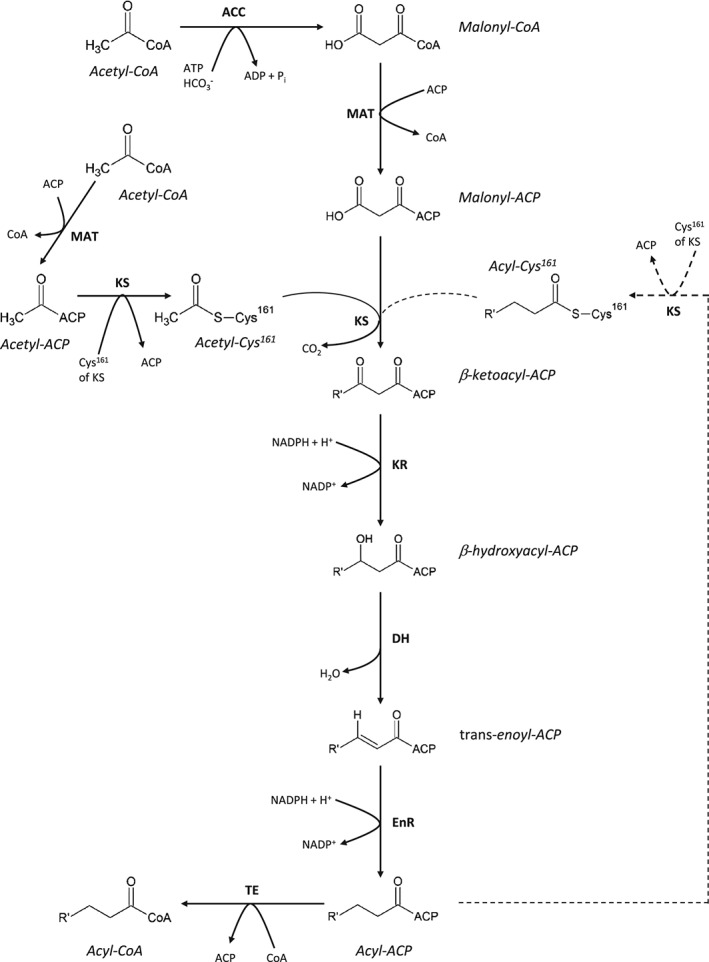
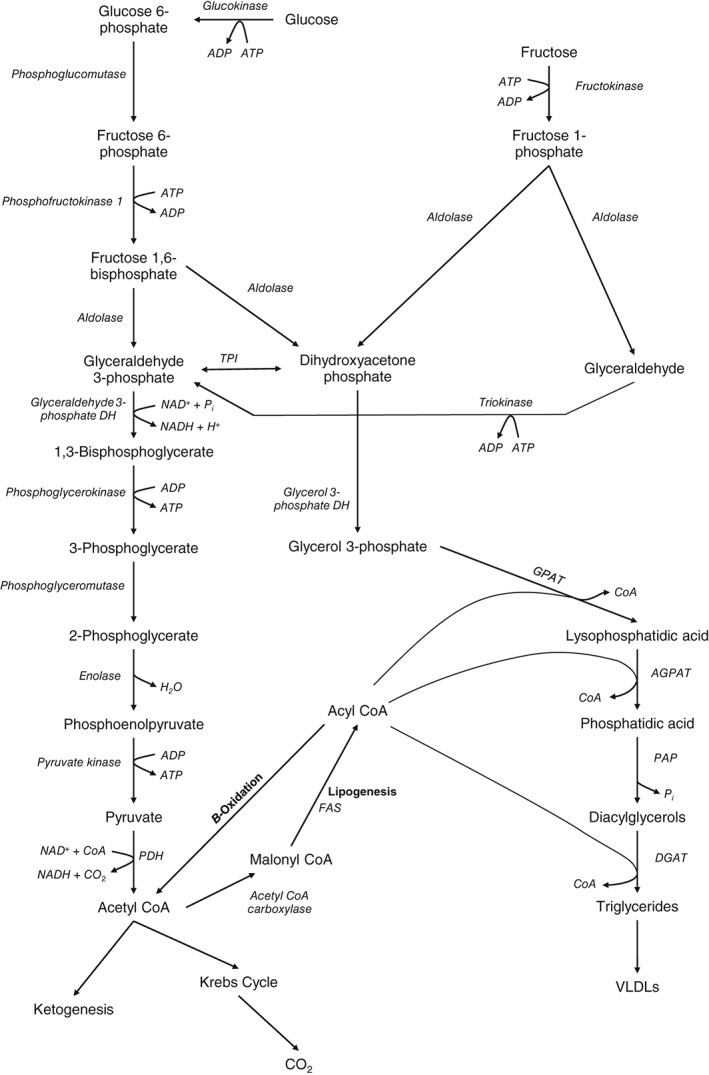
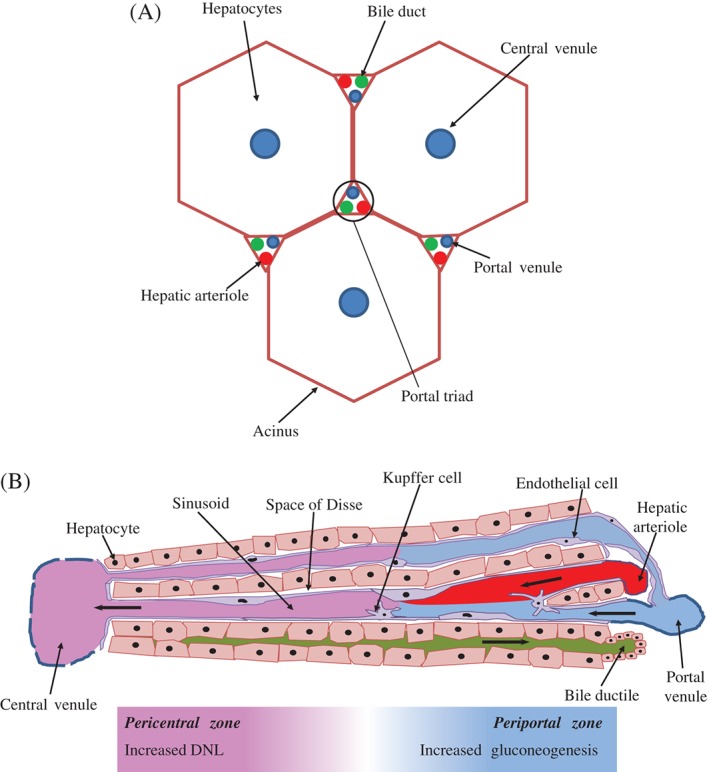
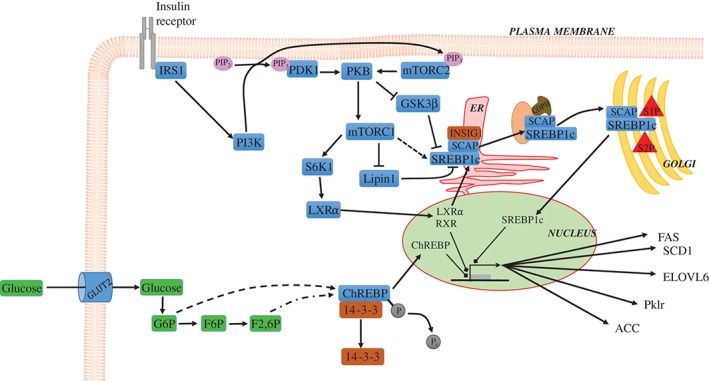
Hepatic de novo lipogenesis (DNL) is the biochemical process of synthesising fatty acids from acetyl-CoA subunits that are produced from a number of different pathways within the cell, most commonly carbohydrate catabolism. In addition to glucose which most commonly supplies carbon units for DNL, fructose is also a profoundly lipogenic substrate that can drive DNL, important when considering the increasing use of fructose in corn syrup as a sweetener. In the context of disease, DNL is thought to contribute to the pathogenesis of non-alcoholic fatty liver disease, a common condition often associated with the metabolic syndrome and consequent insulin resistance. Whether DNL plays a significant role in the pathogenesis of insulin resistance is yet to be fully elucidated, but it may be that the prevalent products of this synthetic process induce some aspect of hepatic insulin resistance.
Keywords: de novo lipogenesis (DNL); fructose; liver; non-alcoholic fatty liver disease (NAFLD); selective insulin resistance.
© 2015 The Authors. Biological Reviews published by John Wiley & Sons Ltd on behalf of Cambridge Philosophical Society.
Figures
References
-
- Abu‐Shanab, A. & Quigley, E. M. M. (2010). The role of the gut microbiota in nonalcoholic fatty liver disease. Nature Reviews Gastroenterology and Hepatology 7, 691–701. - PubMed
-
- Aguado, B. & Campbell, R. D. (1998). Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex. Journal of Biological Chemistry 273, 4096–4105. - PubMed
-
- Ahmad, F. , Ahmad, P. M. , Pieretti, L. & Watters, G. T. (1978). Purification and subunit structure of rat mammary gland acetyl coenzyme A carboxylase. Journal of Biological Chemistry 253, 1733–1737. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
