

PKCη/Rdx-driven phosphorylation of PDK1: a novel mechanism promoting cancer cell survival and permissiveness for parvovirus-induced lysis
- PMID: 25742010
- PMCID: PMC4351090
- DOI: 10.1371/journal.ppat.1004703
PKCη/Rdx-driven phosphorylation of PDK1: a novel mechanism promoting cancer cell survival and permissiveness for parvovirus-induced lysis
Abstract
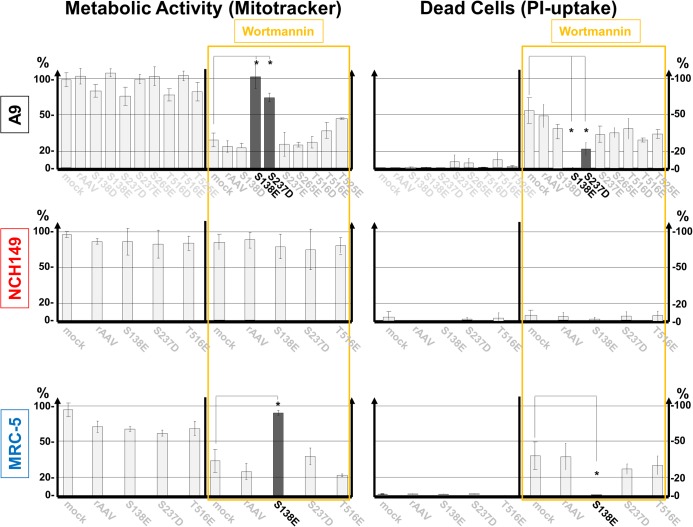
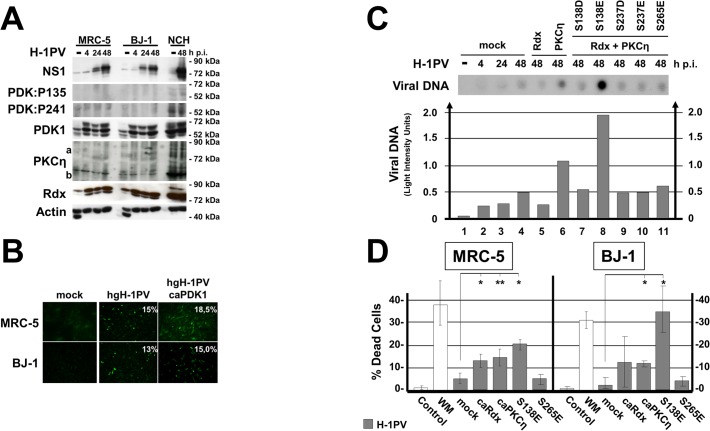
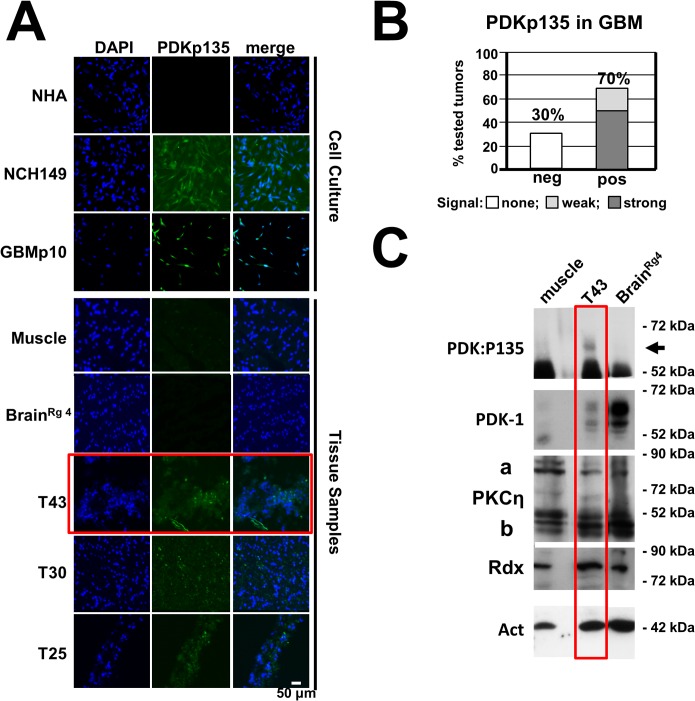
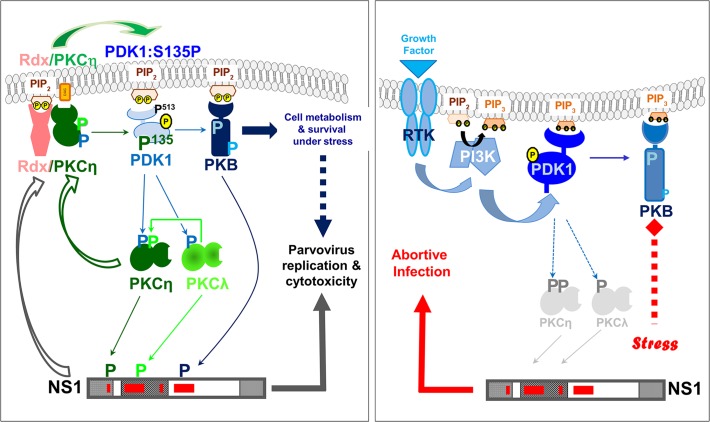
The intrinsic oncotropism and oncosuppressive activities of rodent protoparvoviruses (PVs) are opening new prospects for cancer virotherapy. Virus propagation, cytolytic activity, and spread are tightly connected to activation of the PDK1 signaling cascade, which delays stress-induced cell death and sustains functioning of the parvoviral protein NS1 through PKC(η)-driven modifications. Here we reveal a new PV-induced intracellular loop-back mechanism whereby PKCη/Rdx phosphorylates mouse PDK1:S138 and activates it independently of PI3-kinase signaling. The corresponding human PDK1phosphoS135 appears as a hallmark of highly aggressive brain tumors and may contribute to the very effective targeting of human gliomas by H-1PV. Strikingly, although H-1PV does not trigger PDK1 activation in normal human cells, such cells show enhanced viral DNA amplification and NS1-induced death upon expression of a constitutively active PDK1 mimicking PDK1phosphoS135. This modification thus appears as a marker of human glioma malignant progression and sensitivity to H-1PV-induced tumor cell killing.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Regression of glioma in rat models by intranasal application of parvovirus h-1.Clin Cancer Res. 2011 Aug 15;17(16):5333-42. doi: 10.1158/1078-0432.CCR-10-3124. Epub 2011 Jun 29. Clin Cancer Res. 2011. PMID: 21715567
-
Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models.Neuro Oncol. 2010 Aug;12(8):804-14. doi: 10.1093/neuonc/noq023. Epub 2010 Mar 18. Neuro Oncol. 2010. PMID: 20299703 Free PMC article.
-
Parvovirus interference with intracellular signalling: mechanism of PKCeta activation in MVM-infected A9 fibroblasts.Cell Microbiol. 2008 Mar;10(3):755-69. doi: 10.1111/j.1462-5822.2007.01082.x. Epub 2007 Nov 27. Cell Microbiol. 2008. PMID: 18042254
-
A Roadmap for the Success of Oncolytic Parvovirus-Based Anticancer Therapies.Annu Rev Virol. 2020 Sep 29;7(1):537-557. doi: 10.1146/annurev-virology-012220-023606. Epub 2020 Jun 29. Annu Rev Virol. 2020. PMID: 32600158 Review.
-
Virotherapy of digestive tumors with rodent parvovirus: overview and perspectives.Expert Opin Biol Ther. 2016;16(5):645-53. doi: 10.1517/14712598.2016.1151492. Epub 2016 Mar 28. Expert Opin Biol Ther. 2016. PMID: 26855087 Review.
Cited by
-
Generation and Validation of Monoclonal Antibodies Suitable for Detecting and Monitoring Parvovirus Infections.Pathogens. 2022 Feb 4;11(2):208. doi: 10.3390/pathogens11020208. Pathogens. 2022. PMID: 35215151 Free PMC article.
-
H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future.Viruses. 2019 Jun 18;11(6):562. doi: 10.3390/v11060562. Viruses. 2019. PMID: 31216641 Free PMC article. Review.
-
Oncolytic H-1 parvovirus binds to sialic acid on laminins for cell attachment and entry.Nat Commun. 2021 Jun 22;12(1):3834. doi: 10.1038/s41467-021-24034-7. Nat Commun. 2021. PMID: 34158478 Free PMC article.
-
Role of the phosphatidylinositol-3-kinase/Akt/target of rapamycin pathway during ambidensovirus infection of insect cells.J Gen Virol. 2016 Jan;97(1):233-245. doi: 10.1099/jgv.0.000327. Epub 2015 Oct 26. J Gen Virol. 2016. PMID: 26508507 Free PMC article.
-
Tumor Selectivity of Oncolytic Parvoviruses: From in vitro and Animal Models to Cancer Patients.Front Bioeng Biotechnol. 2015 Apr 22;3:55. doi: 10.3389/fbioe.2015.00055. eCollection 2015. Front Bioeng Biotechnol. 2015. PMID: 25954743 Free PMC article. Review.
References
-
- Hallauer C, Kronauer G, Siegl G (1971) Parvoiruses as contaminants of permanent human cell lines. I. Virus isolation from 1960–1970. Arch Gesamte Virusforsch 35: 80–90. - PubMed
-
- Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, et al. (2012) Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 12: 99 10.1186/1471-2407-12-99 - DOI - PMC - PubMed
-
- Nüesch JPF (2006) Regulation of non-structural protein functions by differential synthesis, modification and trafficking In: Kerr M.E., CSB; Linden R.M.; Parrish C.R.; Cotmore S.F., editor. Parvoviruses. London: Edward Arnold, Ltd. pp. 275–290.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
