

Comparative genomics of Cluster O mycobacteriophages
- PMID: 25742016
- PMCID: PMC4351075
- DOI: 10.1371/journal.pone.0118725
Comparative genomics of Cluster O mycobacteriophages
Abstract
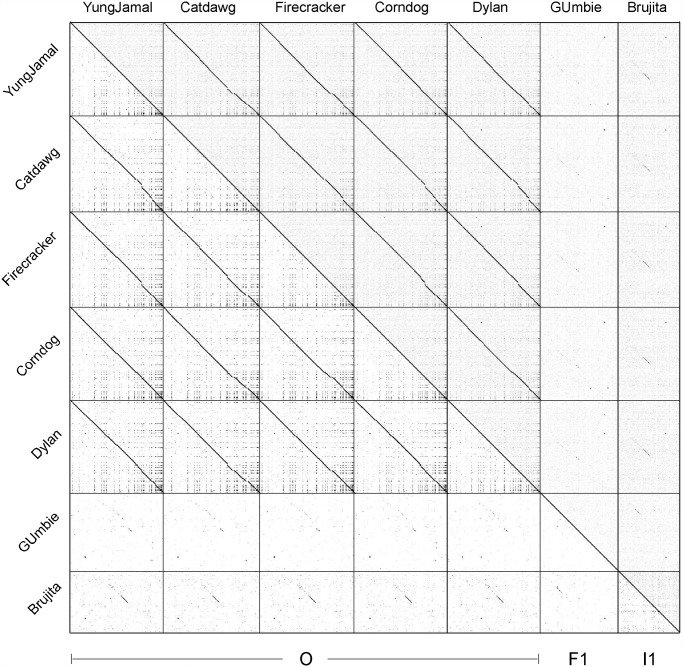
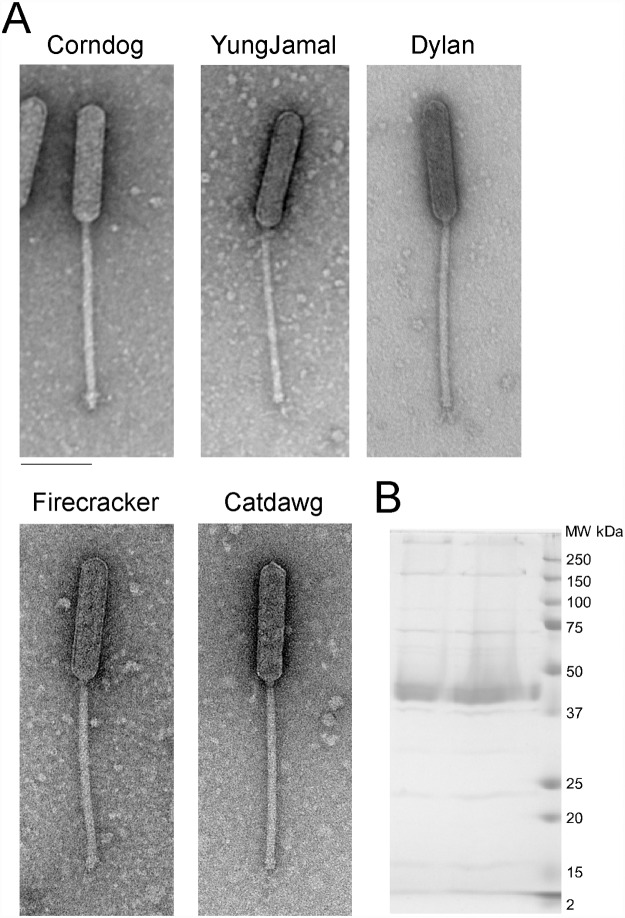
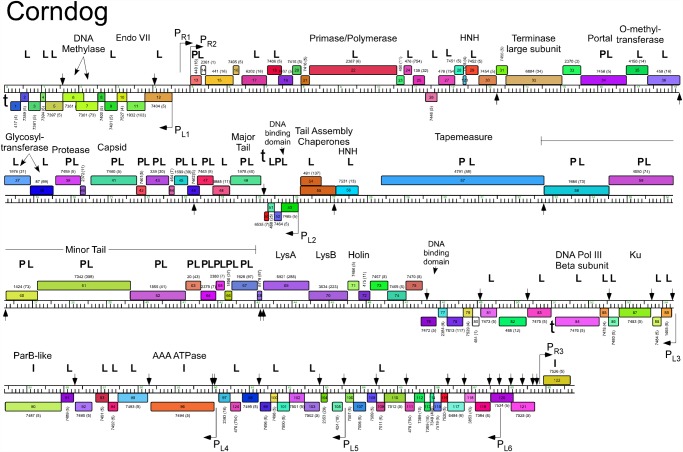
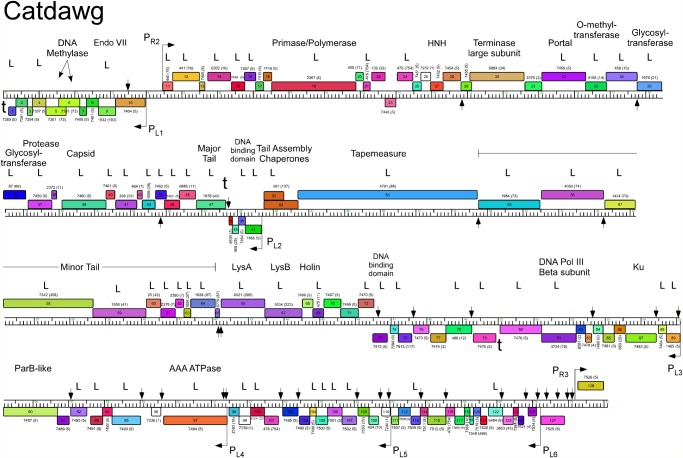
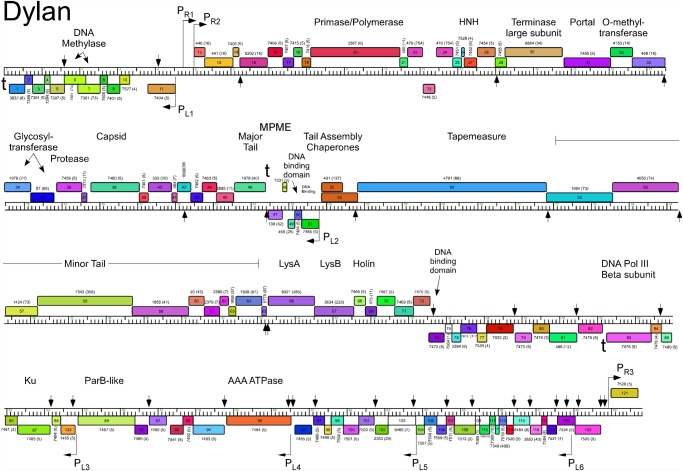
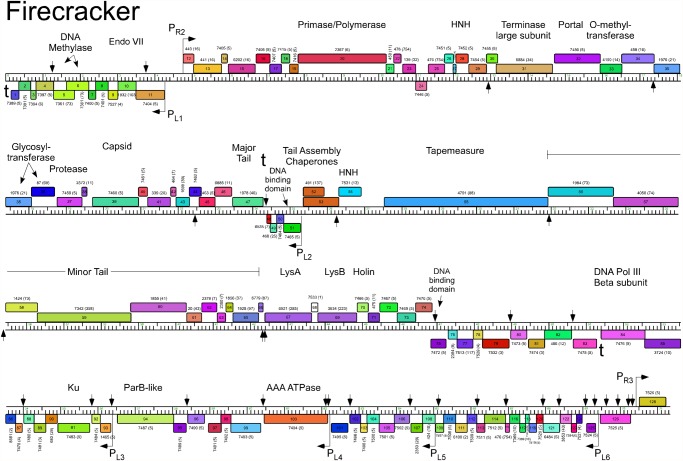
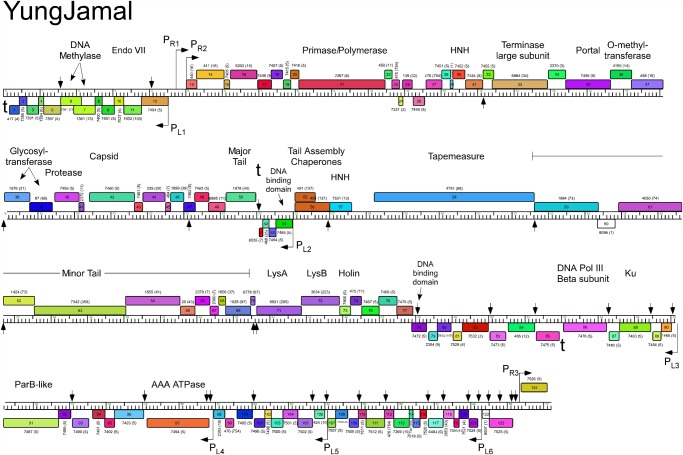
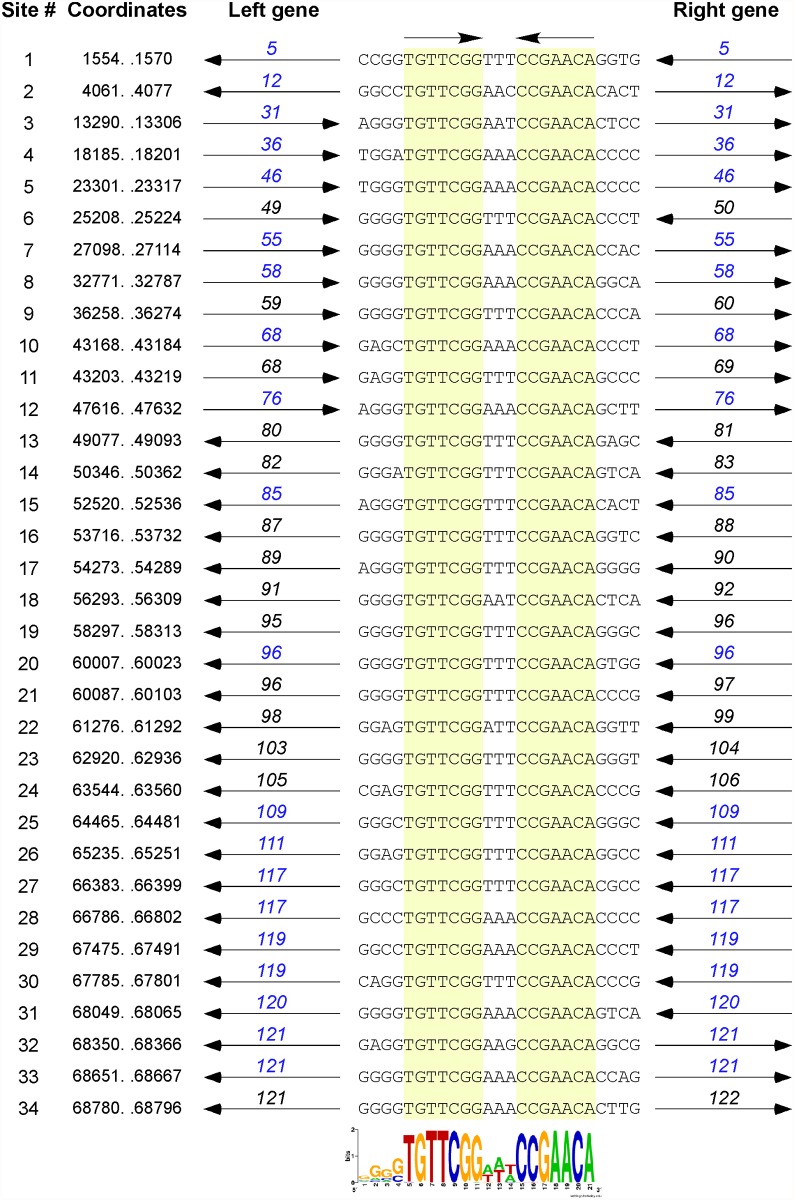
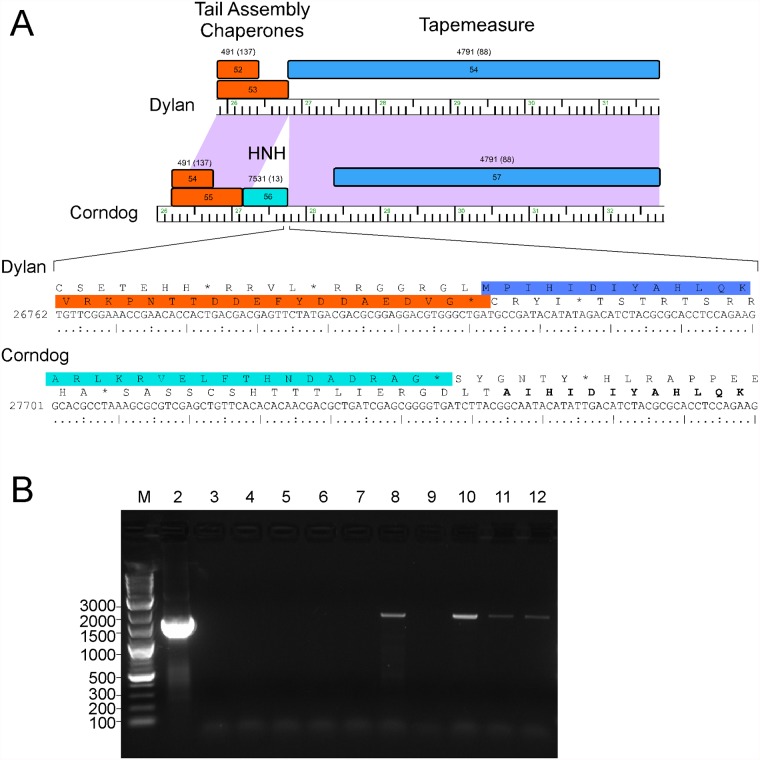
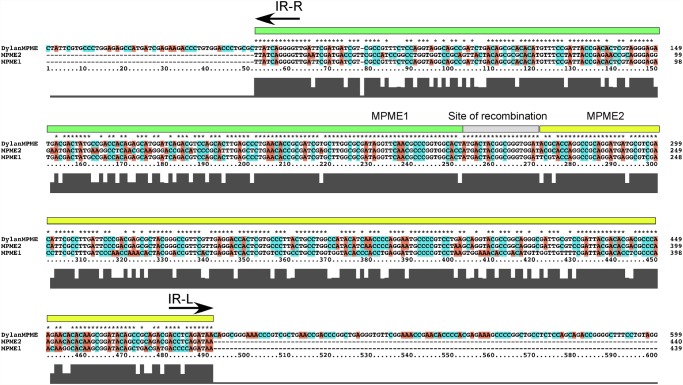
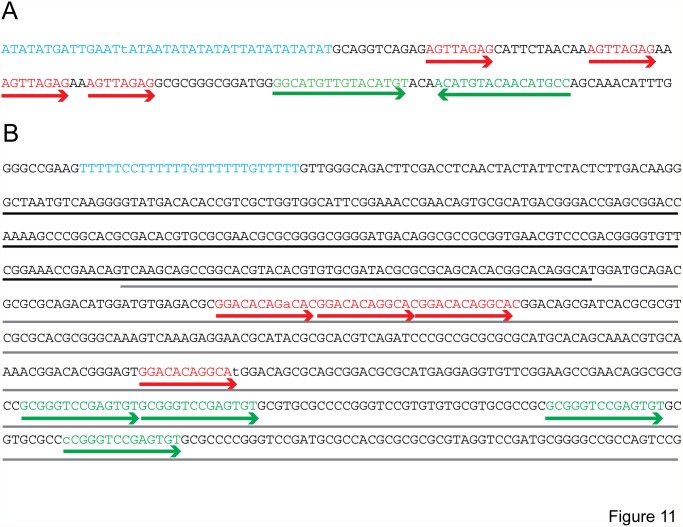
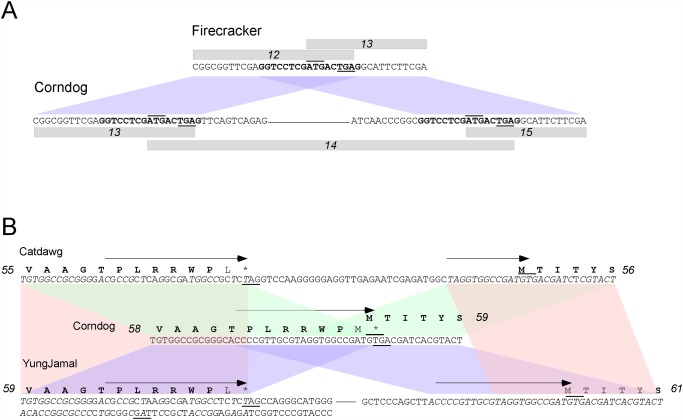
Mycobacteriophages--viruses of mycobacterial hosts--are genetically diverse but morphologically are all classified in the Caudovirales with double-stranded DNA and tails. We describe here a group of five closely related mycobacteriophages--Corndog, Catdawg, Dylan, Firecracker, and YungJamal--designated as Cluster O with long flexible tails but with unusual prolate capsids. Proteomic analysis of phage Corndog particles, Catdawg particles, and Corndog-infected cells confirms expression of half of the predicted gene products and indicates a non-canonical mechanism for translation of the Corndog tape measure protein. Bioinformatic analysis identifies 8-9 strongly predicted SigA promoters and all five Cluster O genomes contain more than 30 copies of a 17 bp repeat sequence with dyad symmetry located throughout the genomes. Comparison of the Cluster O phages provides insights into phage genome evolution including the processes of gene flux by horizontal genetic exchange.
Conflict of interest statement
Figures
References
-
- Hendrix RW. Bacteriophages In: Knipe DM, Howley PM, editors. Fields Virology, Sixth Edition Philadelphia: Lippincott Williams & Wilkins; 2013.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
