

An XRCC4 splice mutation associated with severe short stature, gonadal failure, and early-onset metabolic syndrome
- PMID: 25742519
- PMCID: PMC4422886
- DOI: 10.1210/jc.2015-1098
An XRCC4 splice mutation associated with severe short stature, gonadal failure, and early-onset metabolic syndrome
Abstract
Context: Severe short stature can be caused by defects in numerous biological processes including defects in IGF-1 signaling, centromere function, cell cycle control, and DNA damage repair. Many syndromic causes of short stature are associated with medical comorbidities including hypogonadism and microcephaly.
Objective: To identify an underlying genetic etiology in two siblings with severe short stature and gonadal failure.
Design: Clinical phenotyping, genetic analysis, complemented by in vitro functional studies of the candidate gene.
Setting: An academic pediatric endocrinology clinic.
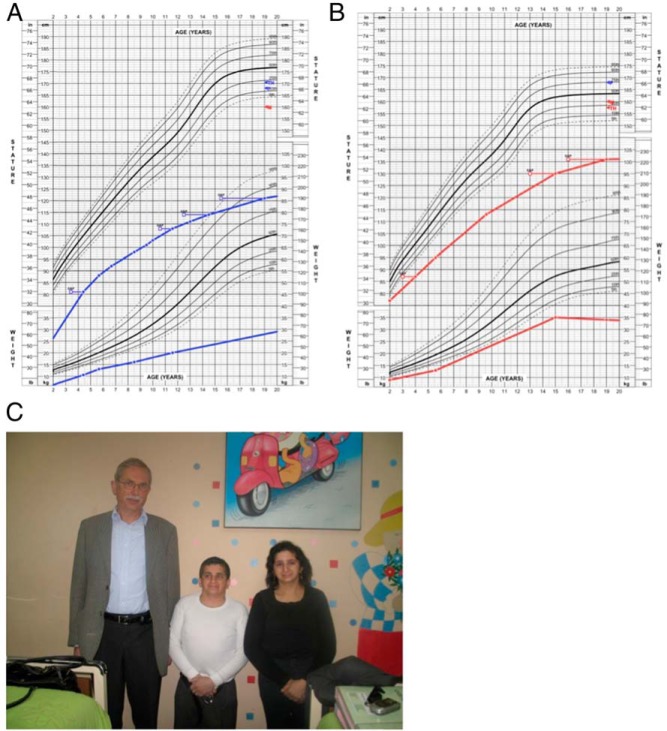
Patients or other participants: Two adult siblings (male patient [P1] and female patient 2 [P2]) presented with a history of severe postnatal growth failure (adult heights: P1, -6.8 SD score; P2, -4 SD score), microcephaly, primary gonadal failure, and early-onset metabolic syndrome in late adolescence. In addition, P2 developed a malignant gastrointestinal stromal tumor at age 28.
Intervention(s): Single nucleotide polymorphism microarray and exome sequencing.
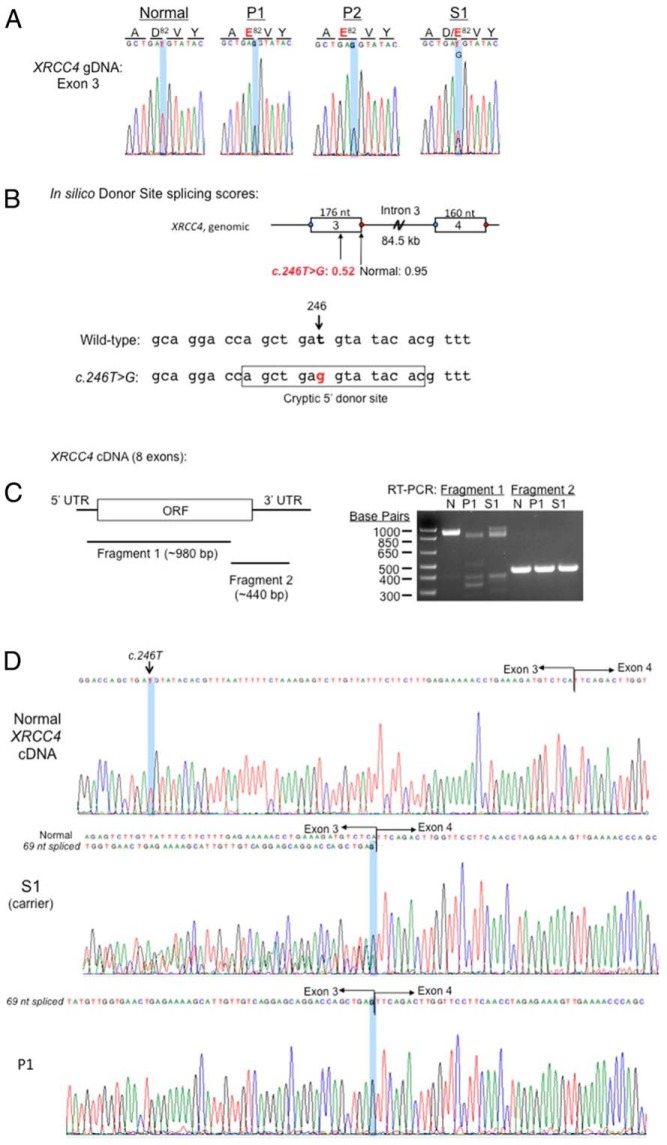
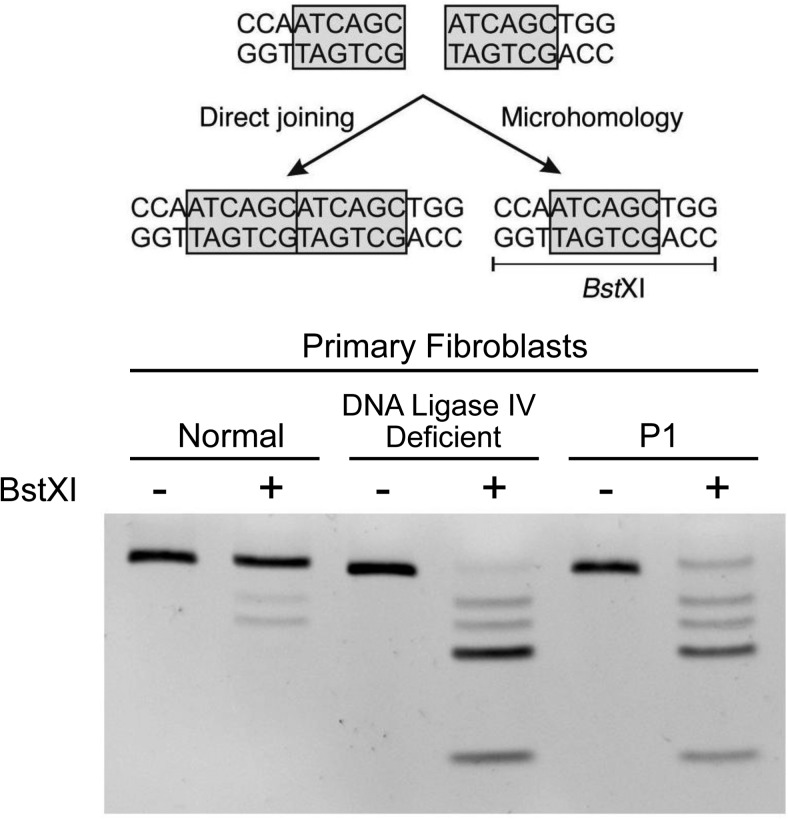
Results: Combined microarray analysis and whole exome sequencing of the two affected siblings and one unaffected sister identified a homozygous variant in XRCC4 as the probable candidate variant. Sanger sequencing and mRNA studies revealed a splice variant resulting in an in-frame deletion of 23 amino acids. Primary fibroblasts (P1) showed a DNA damage repair defect.
Conclusions: In this study we have identified a novel pathogenic variant in XRCC4, a gene that plays a critical role in non-homologous end-joining DNA repair. This finding expands the spectrum of DNA damage repair syndromes to include XRCC4 deficiency causing severe postnatal growth failure, microcephaly, gonadal failure, metabolic syndrome, and possibly tumor predisposition.
Figures
References
-
- Bicknell LS, Walker S, Klingseisen A, et al. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet. 2011;43:350–355. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
