

Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation
- PMID: 25742700
- PMCID: PMC4400290
- DOI: 10.1182/blood-2014-12-619783
Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation
Abstract
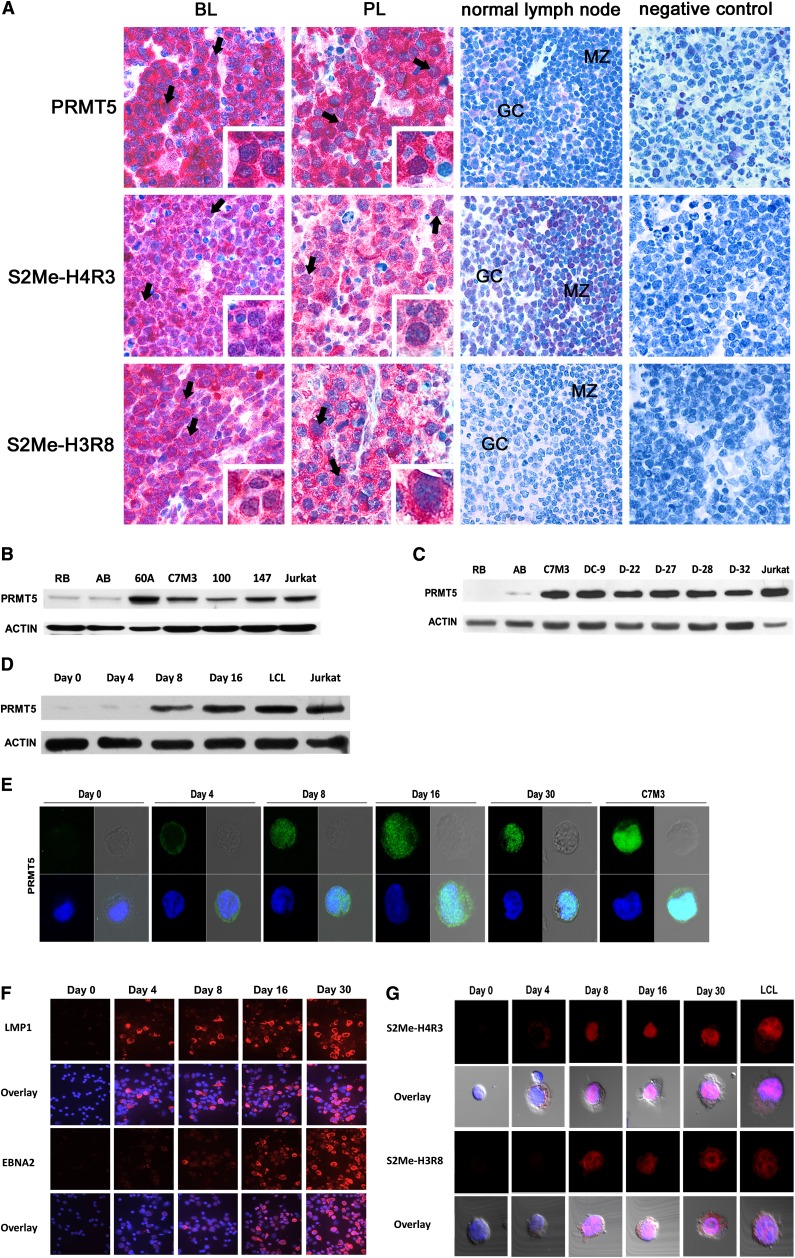
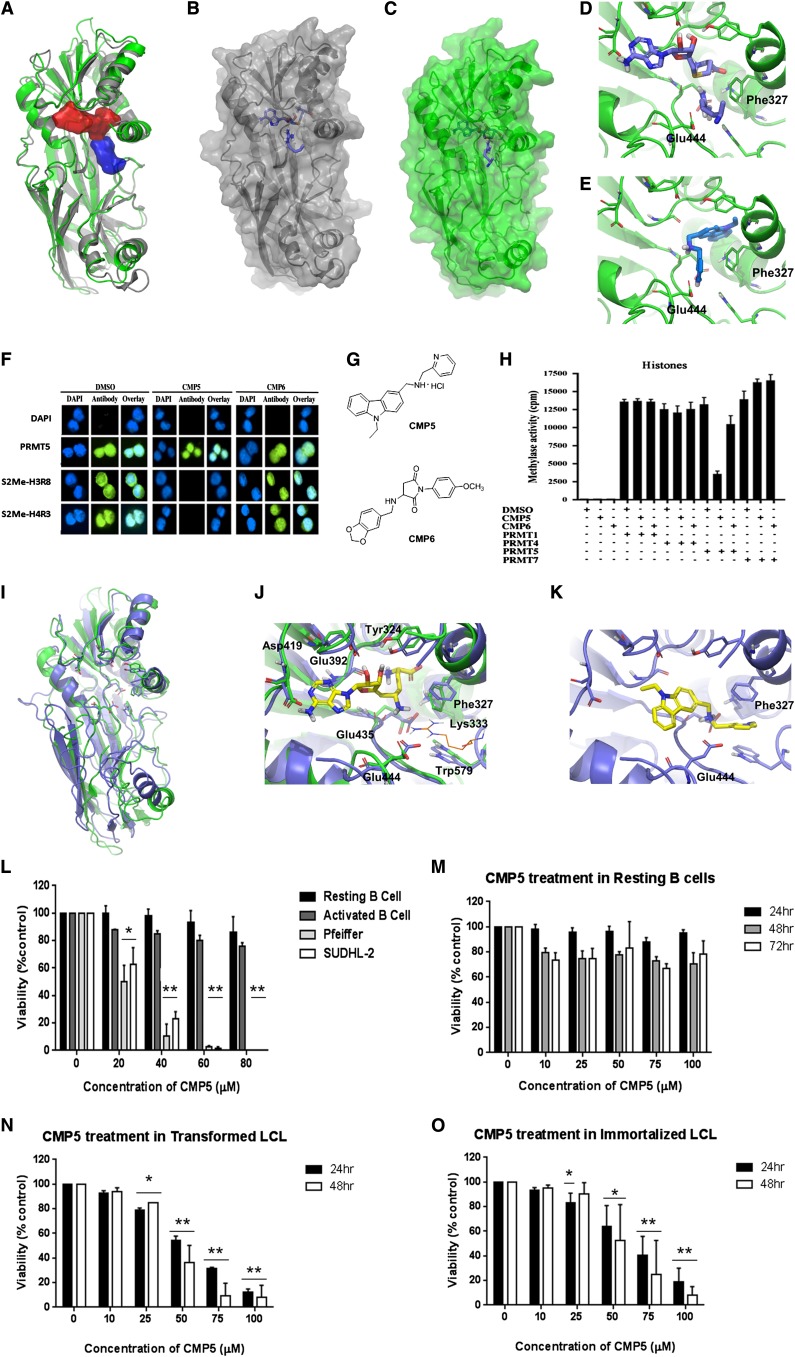
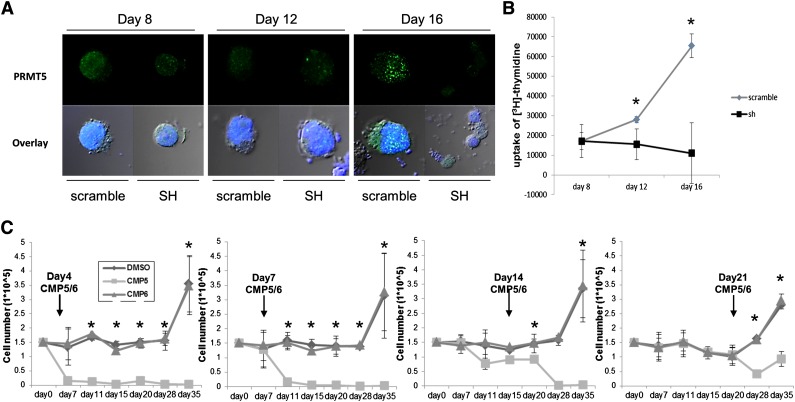
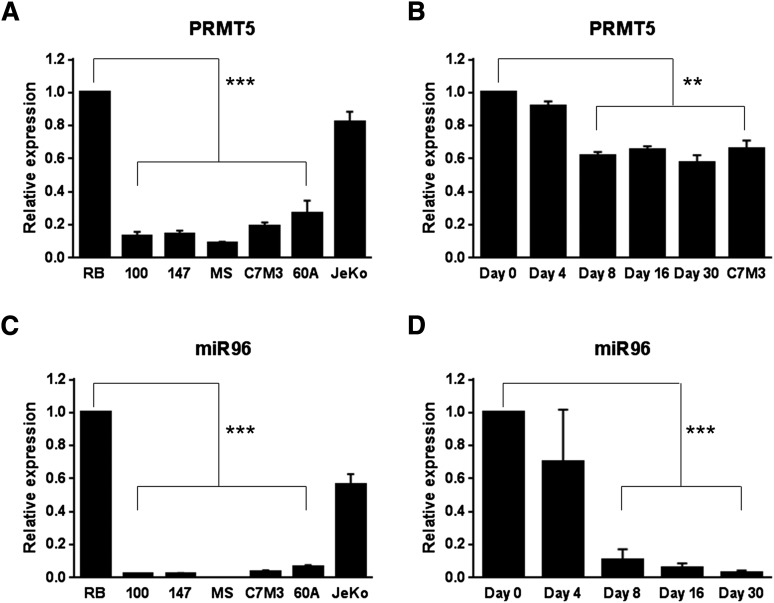
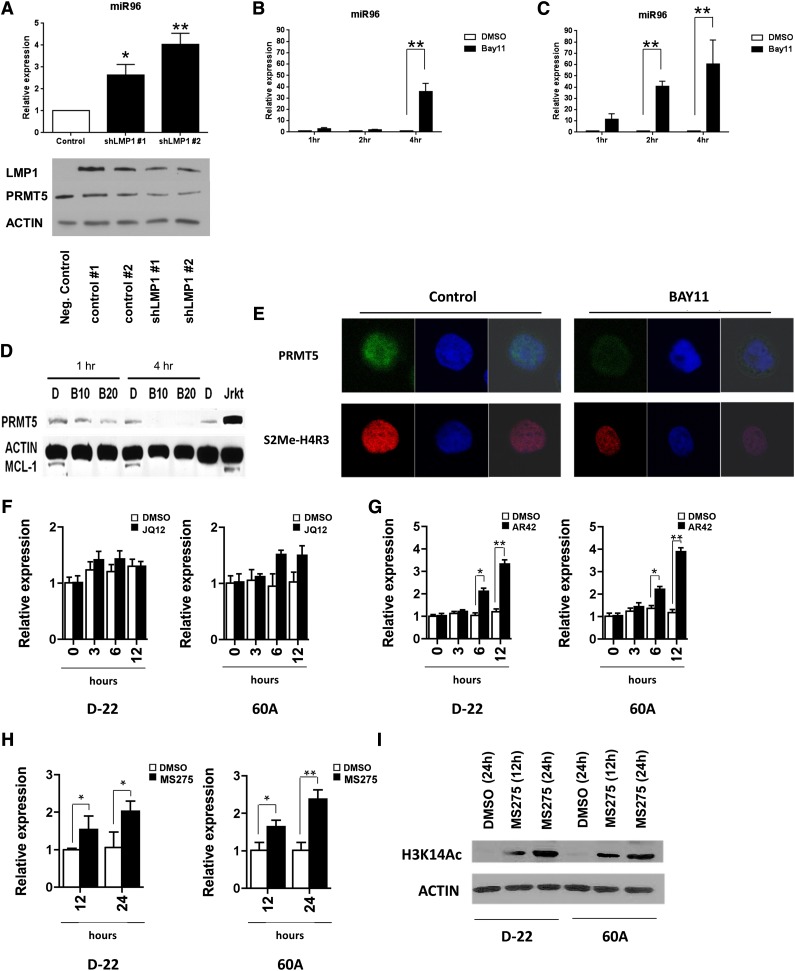
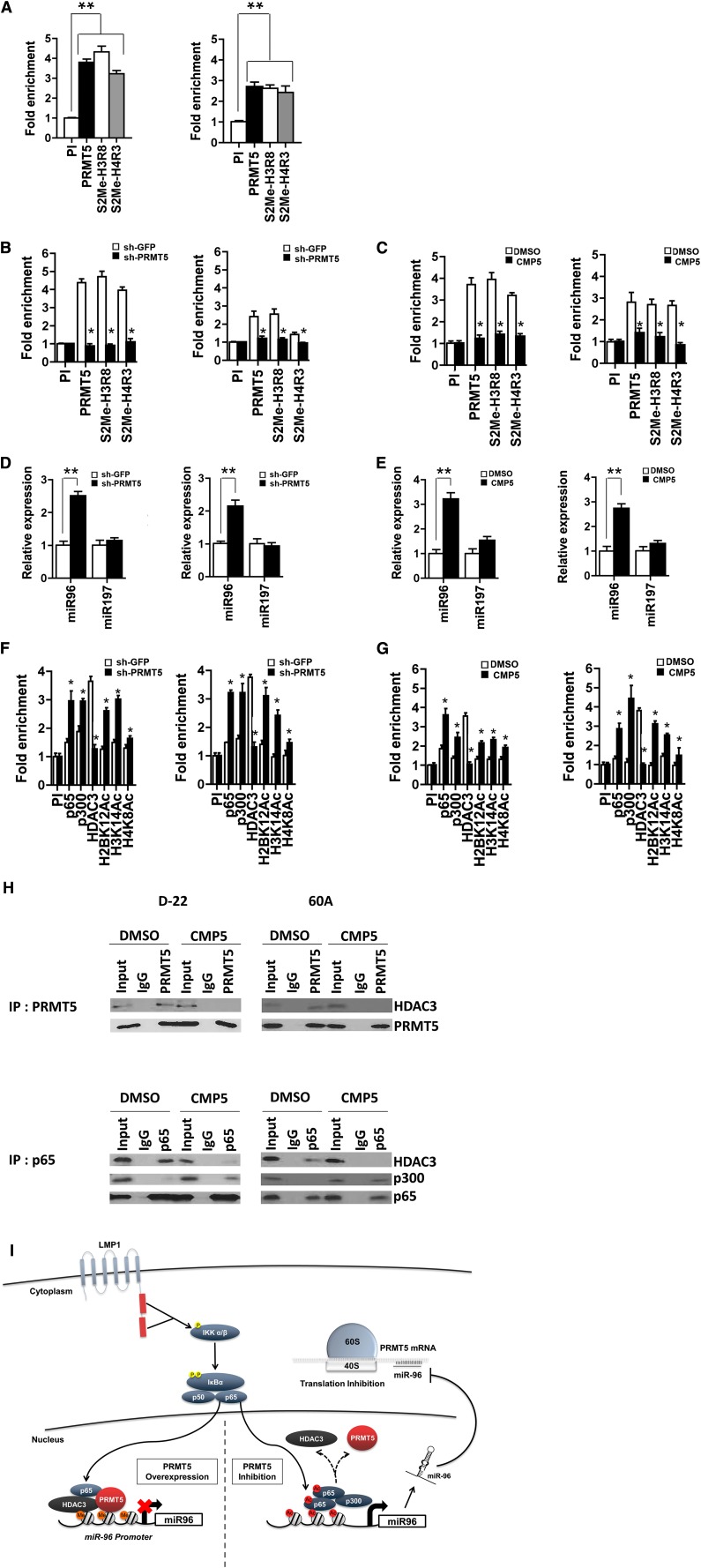
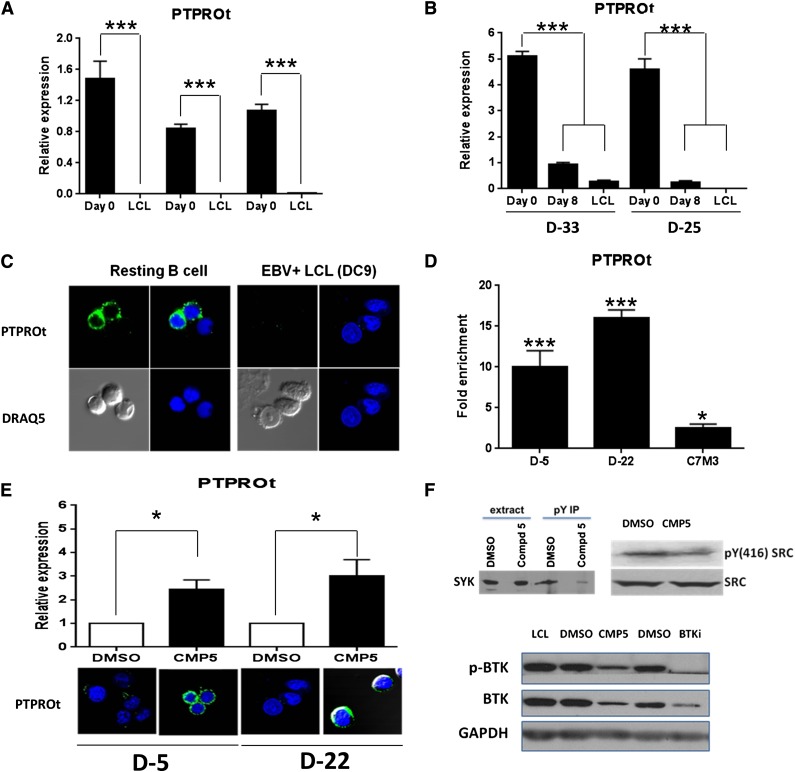
Epigenetic events that are essential drivers of lymphocyte transformation remain incompletely characterized. We used models of Epstein-Barr virus (EBV)-induced B-cell transformation to document the relevance of protein arginine methyltransferase 5 (PRMT5) to regulation of epigenetic-repressive marks during lymphomagenesis. EBV(+) lymphomas and transformed cell lines exhibited abundant expression of PRMT5, a type II PRMT enzyme that promotes transcriptional silencing of target genes by methylating arginine residues on histone tails. PRMT5 expression was limited to EBV-transformed cells, not resting or activated B lymphocytes, validating it as an ideal therapeutic target. We developed a first-in-class, small-molecule PRMT5 inhibitor that blocked EBV-driven B-lymphocyte transformation and survival while leaving normal B cells unaffected. Inhibition of PRMT5 led to lost recruitment of a PRMT5/p65/HDAC3-repressive complex on the miR96 promoter, restored miR96 expression, and PRMT5 downregulation. RNA-sequencing and chromatin immunoprecipitation experiments identified several tumor suppressor genes, including the protein tyrosine phosphatase gene PTPROt, which became silenced during EBV-driven B-cell transformation. Enhanced PTPROt expression following PRMT5 inhibition led to dephosphorylation of kinases that regulate B-cell receptor signaling. We conclude that PRMT5 is critical to EBV-driven B-cell transformation and maintenance of the malignant phenotype, and that PRMT5 inhibition shows promise as a novel therapeutic approach for B-cell lymphomas.
© 2015 by The American Society of Hematology.
Figures
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. - PubMed
-
- Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4(10):757–768. - PubMed
-
- Jansson M, Durant ST, Cho EC, et al. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10(12):1431–1439. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
