

Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate
- PMID: 25742913
- PMCID: PMC4415062
- DOI: 10.1093/cvr/cvv097
Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate
Abstract
Aims: The mitochondrial permeability transition pore (mPTP) plays a central role for tissue damage and cell death during ischaemia-reperfusion (I/R). We investigated the contribution of mitochondrial inorganic polyphosphate (polyP), a potent activator of Ca(2+)-induced mPTP opening, towards mPTP activation and cardiac cell death in I/R.
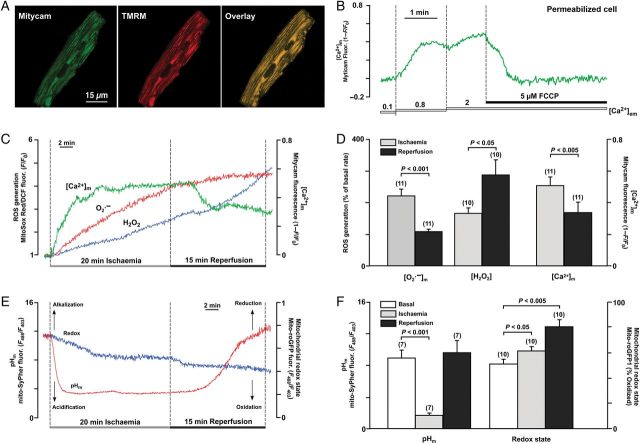
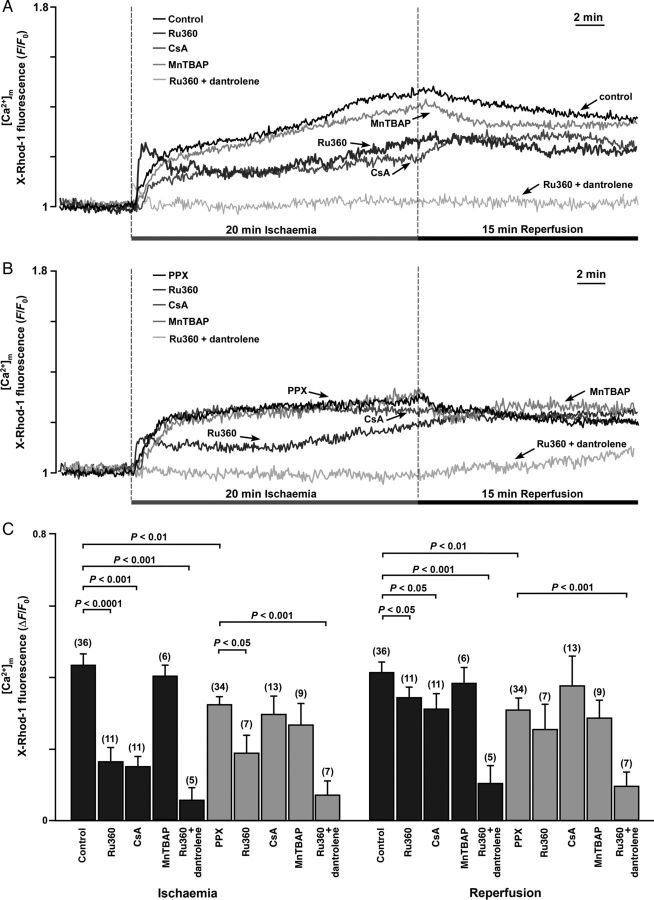
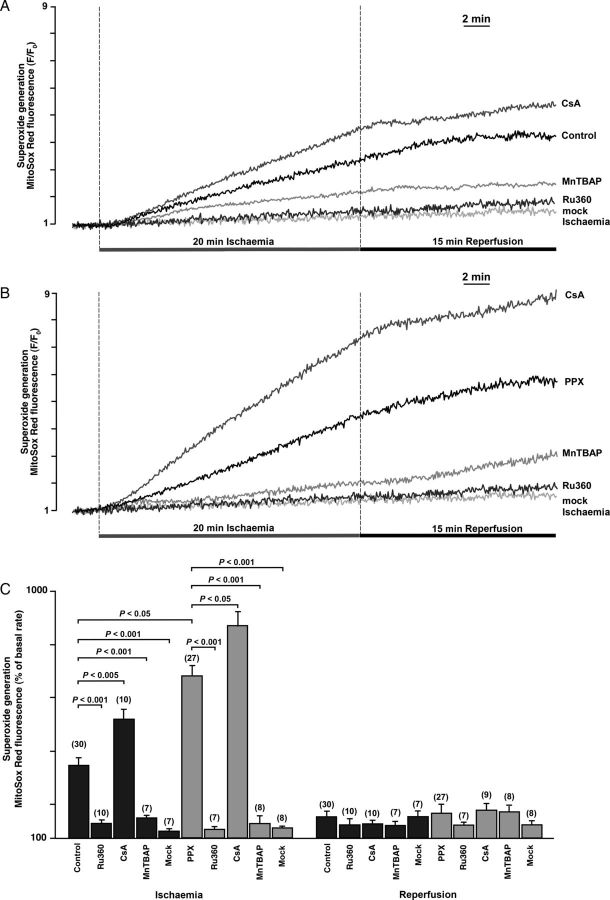
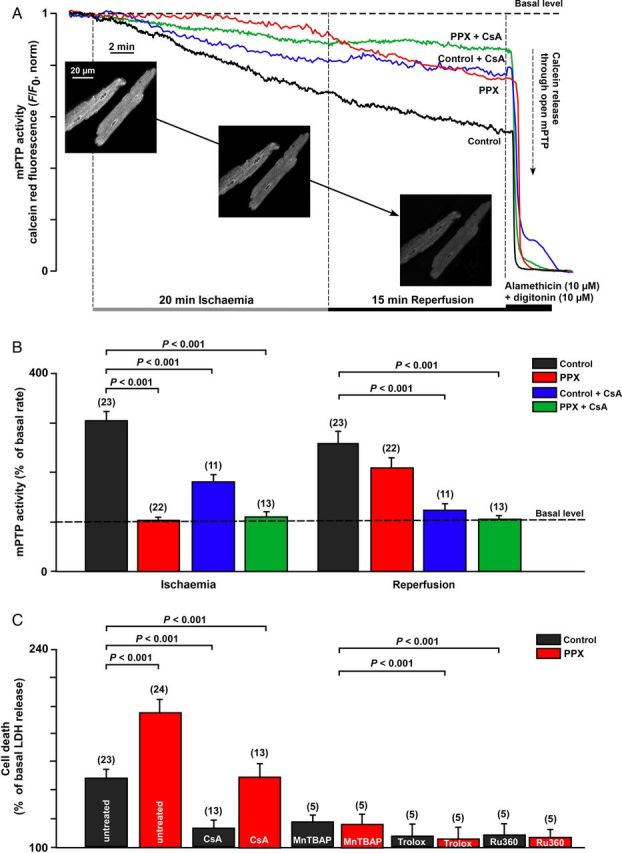
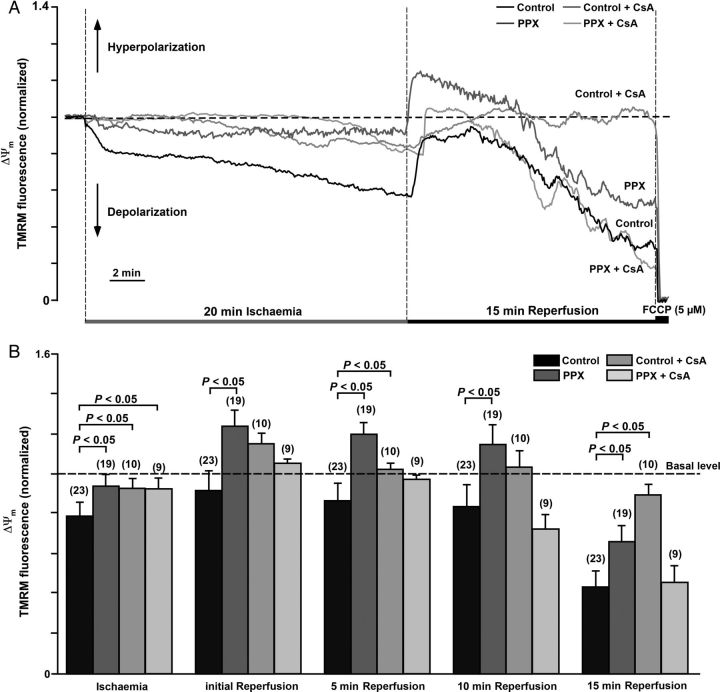
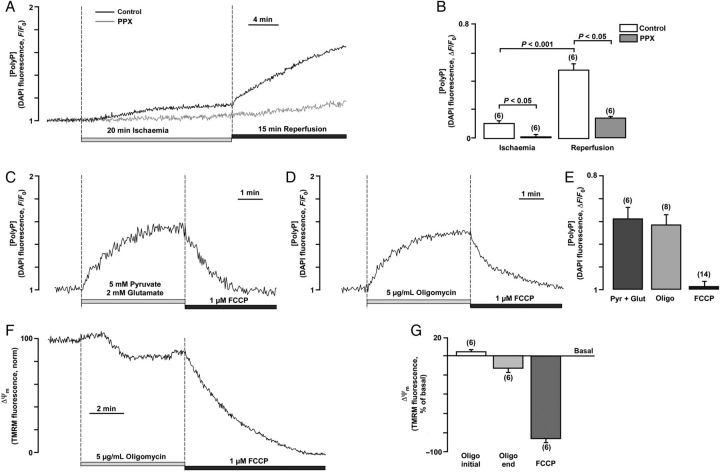
Methods and results: A significant increase in mitochondrial free calcium concentration ([Ca(2+)]m), reactive oxygen species (ROS) generation, mitochondrial membrane potential depolarization (ΔΨm), and mPTP activity, but no cell death, was observed after 20 min of ischaemia. The [Ca(2+)]m increase during ischaemia was partially prevented by the mitochondrial Ca(2+) uniporter (MCU) inhibitor Ru360 and completely abolished by the combination of Ru360 and the ryanodine receptor type 1 blocker dantrolene, suggesting two complimentary Ca(2+) uptake mechanisms. In the absence of Ru360 and dantrolene, mPTP closing by polyP depletion or CSA decreased mitochondrial Ca(2+) uptake, suggesting that during ischaemia Ca(2+) can enter mitochondria through mPTP. During reperfusion, a burst of endogenous polyP production coincided with a decrease in [Ca(2+)]m, a decline in superoxide generation, and an acceleration of hydrogen peroxide (H2O2) production. An increase in H2O2 correlated with restoration of mitochondrial pHm and an increase in cell death. mPTP opening and cell death on reperfusion were prevented by antioxidants Trolox and MnTBAP [Mn (III) tetrakis (4-benzoic acid) porphyrin chloride]. Enzymatic polyP depletion did not affect mPTP opening during reperfusion, but increased ROS generation and cell death, suggesting that polyP plays a protective role in cellular stress response.
Conclusions: Transient Ca(2+)/polyP-mediated mPTP opening during ischaemia may serve to protect cells against cytosolic Ca(2+) overload, whereas ROS/pH-mediated sustained mPTP opening on reperfusion induces cell death.
Keywords: Inorganic polyphosphate; Ischaemia–reperfusion injury; Mitochondrial permeability transition pore; Mitochondrial ryanodine receptor; Oxidative stress.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2015. For permissions please email: journals.permissions@oup.com.
Figures
References
-
- Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 2015;78:129–141. - PubMed
-
- Halestrap AP. Mitochondria and reperfusion injury of the heart—a holey death but not beyond salvation. J Bioenerg Biomembr 2009;41:113–121. - PubMed
-
- Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem 2001;276:12030–12034. - PubMed
-
- Di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett 2001;492:4–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
