

Genomic landscape of paediatric adrenocortical tumours
- PMID: 25743702
- PMCID: PMC4352712
- DOI: 10.1038/ncomms7302
Genomic landscape of paediatric adrenocortical tumours
Abstract
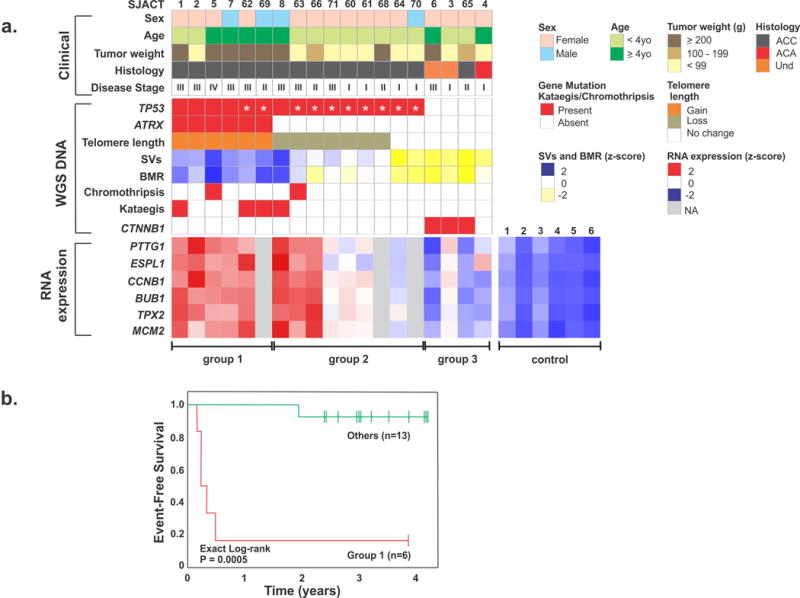
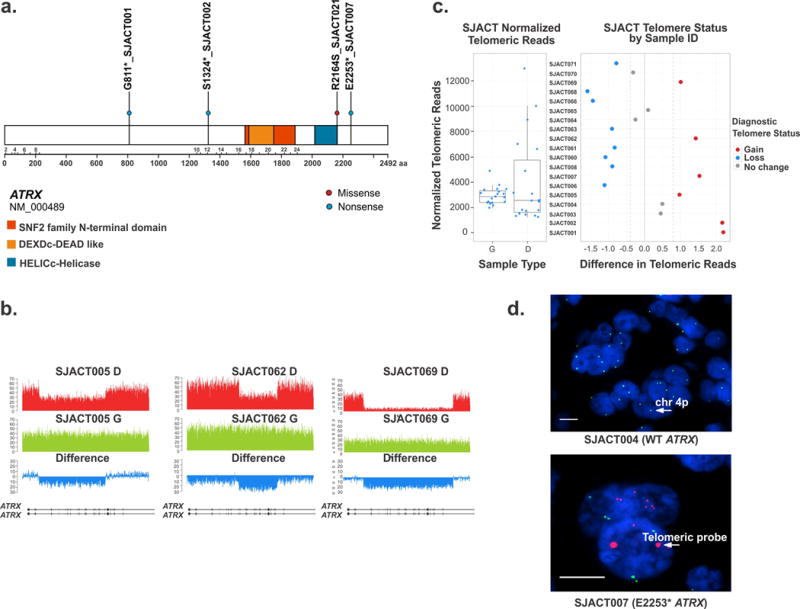
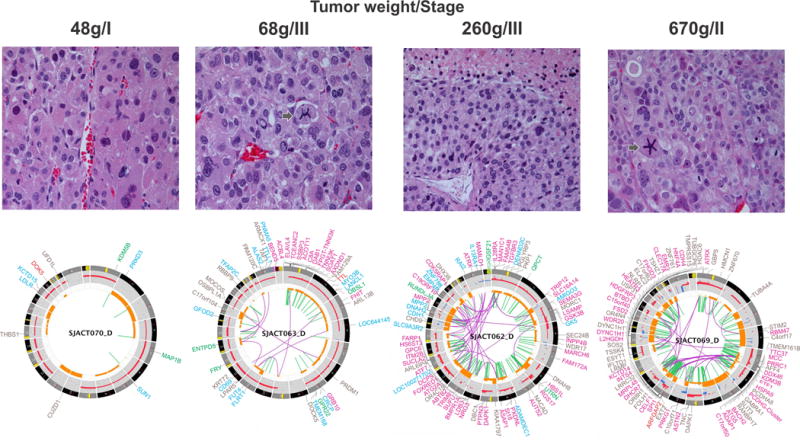
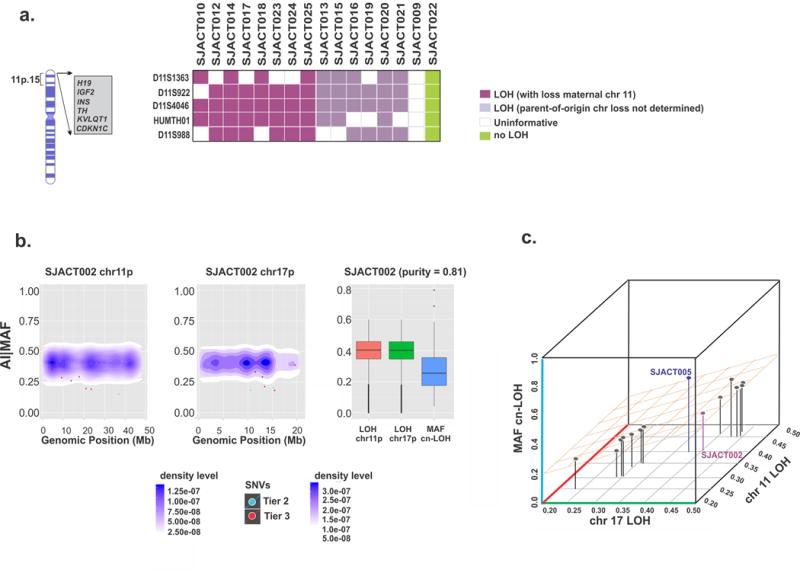
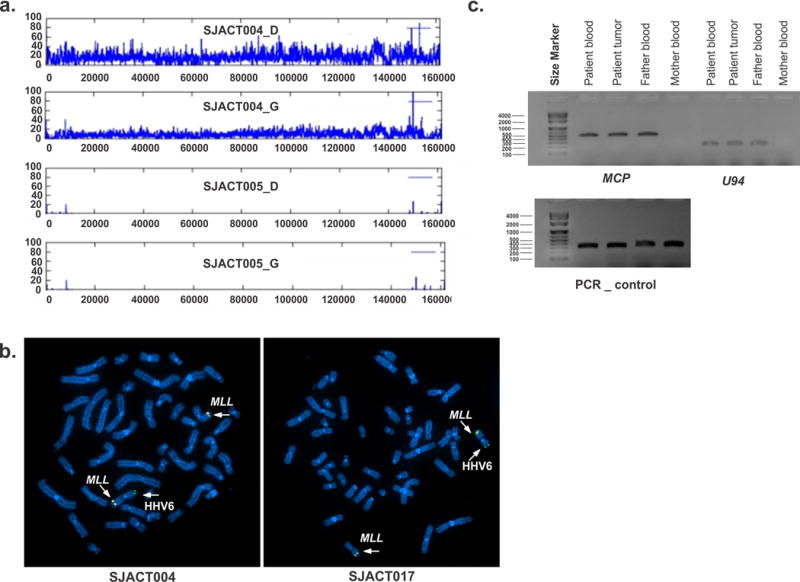
Paediatric adrenocortical carcinoma is a rare malignancy with poor prognosis. Here we analyse 37 adrenocortical tumours (ACTs) by whole-genome, whole-exome and/or transcriptome sequencing. Most cases (91%) show loss of heterozygosity (LOH) of chromosome 11p, with uniform selection against the maternal chromosome. IGF2 on chromosome 11p is overexpressed in 100% of the tumours. TP53 mutations and chromosome 17 LOH with selection against wild-type TP53 are observed in 28 ACTs (76%). Chromosomes 11p and 17 undergo copy-neutral LOH early during tumorigenesis, suggesting tumour-driver events. Additional genetic alterations include recurrent somatic mutations in ATRX and CTNNB1 and integration of human herpesvirus-6 in chromosome 11p. A dismal outcome is predicted by concomitant TP53 and ATRX mutations and associated genomic abnormalities, including massive structural variations and frequent background mutations. Collectively, these findings demonstrate the nature, timing and potential prognostic significance of key genetic alterations in paediatric ACT and outline a hypothetical model of paediatric adrenocortical tumorigenesis.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Ribeiro RC, Pinto EM, Zambetti GP, Rodriguez-Galindo C. The International Pediatric Adrenocortical Tumor Registry initiative: contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol. 2012;351:37–43. - PubMed
-
- Michalkiewicz E, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22:838–845. - PubMed
-
- Steenman M, Westerveld A, Mannens M. Genetics of Beckwith-Wiedemann syndrome-associated tumors: common genetic pathways. Genes Chromosom Cancer. 2000;28:1–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
