

Genetic Ablation of PDGF-Dependent Signaling Pathways Abolishes Vascular Remodeling and Experimental Pulmonary Hypertension
- PMID: 25745058
- PMCID: PMC5588904
- DOI: 10.1161/ATVBAHA.114.304864
Genetic Ablation of PDGF-Dependent Signaling Pathways Abolishes Vascular Remodeling and Experimental Pulmonary Hypertension
Abstract
Objective: Despite modern therapies, pulmonary arterial hypertension (PAH) harbors a high mortality. Vascular remodeling is a hallmark of the disease. Recent clinical studies revealed that antiremodeling approaches with tyrosine-kinase inhibitors such as imatinib are effective, but its applicability is limited by significant side effects. Although imatinib has multiple targets, expression analyses support a role for platelet-derived growth factor (PDGF) in the pathobiology of the disease. However, its precise role and downstream signaling events have not been established.
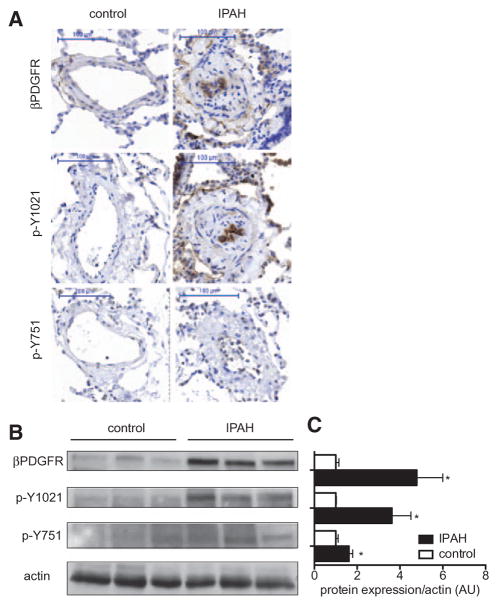
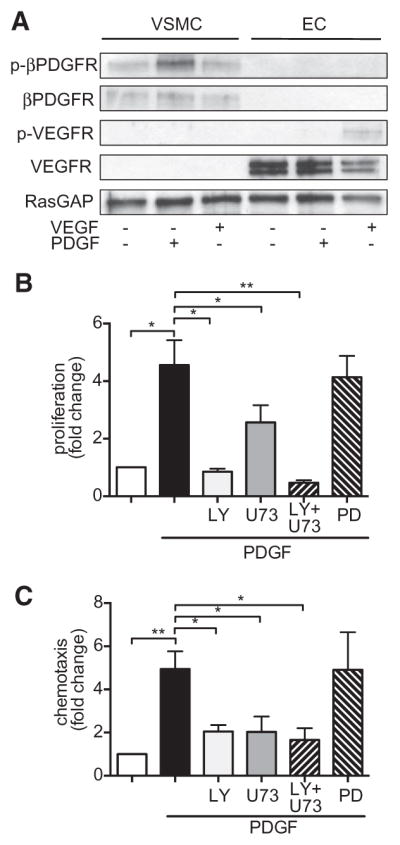
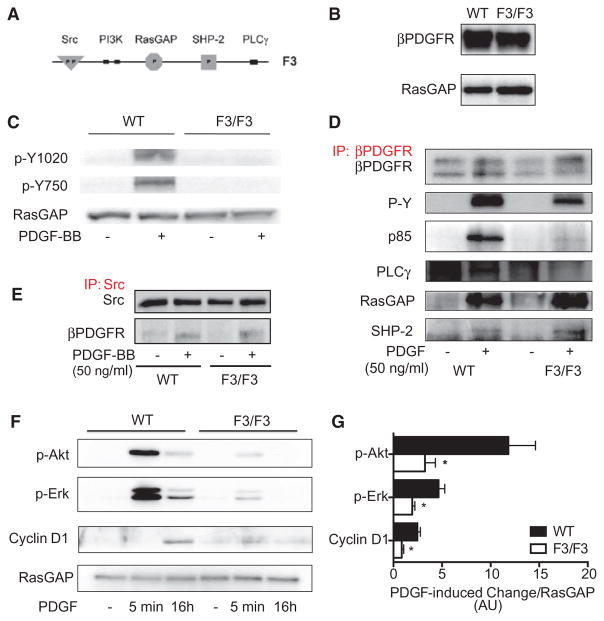
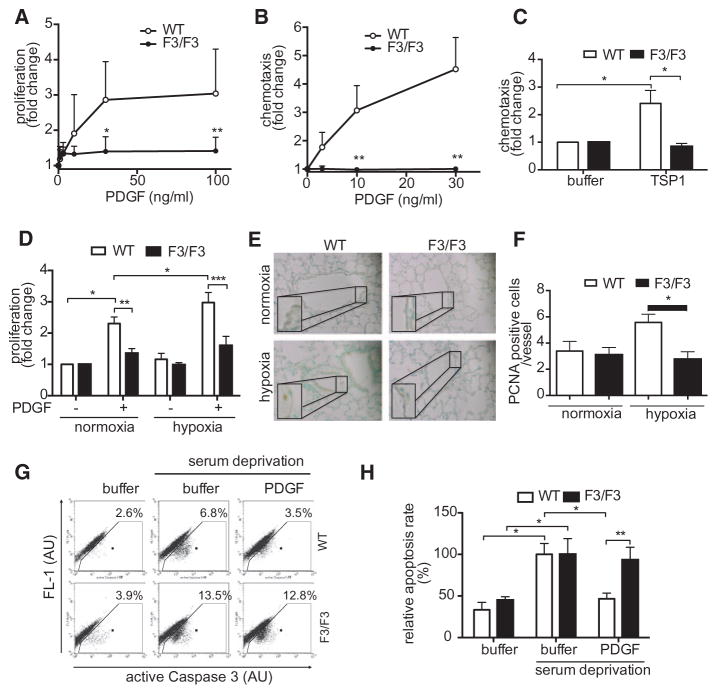
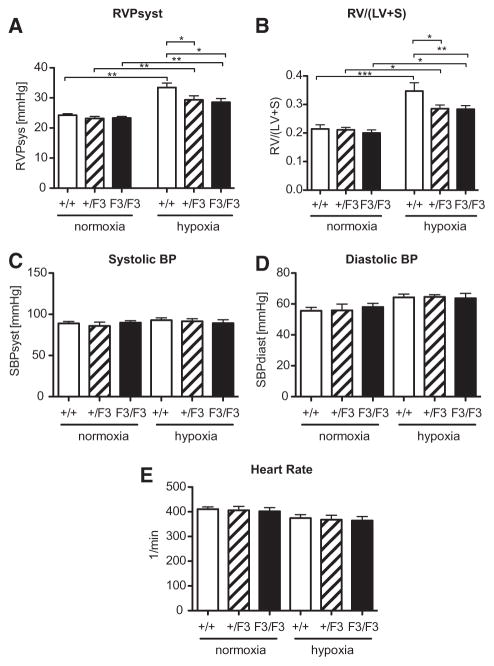
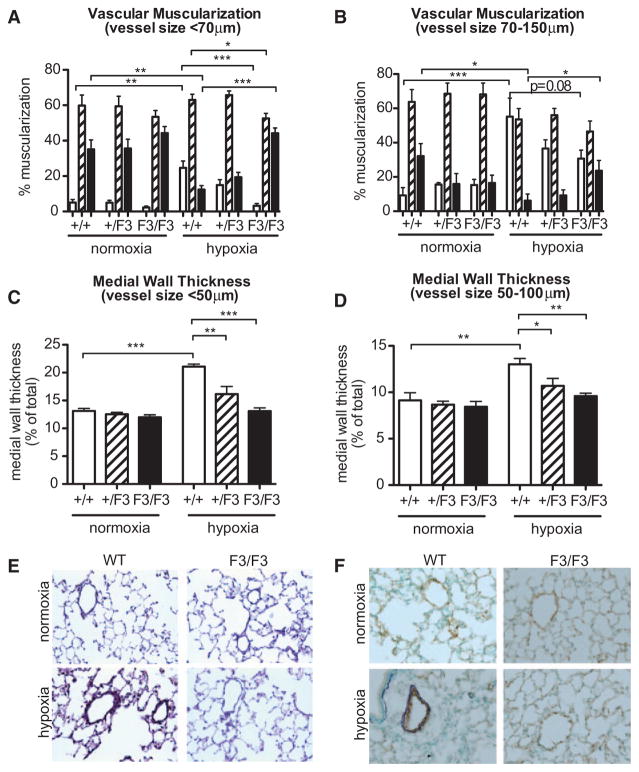
Approach and results: Patients with PAH exhibit enhanced expression and phosphorylation of β PDGF receptor (βPDGFR) in remodeled pulmonary arterioles, particularly at the binding sites for phophatidyl-inositol-3-kinase and PLCγ at tyrosine residues 751 and 1021, respectively. These signaling molecules were identified as critical downstream mediators of βPDGFR-mediated proliferation and migration of pulmonary arterial smooth muscle cells. We, therefore, investigated mice expressing a mutated βPDGFR that is unable to recruit phophatidyl-inositol-3-kinase and PLCγ (βPDGFR(F3/F3)). PDGF-dependent Erk1/2 and Akt phosphorylation, cyclin D1 induction, and proliferation, migration, and protection against apoptosis were abolished in βPDGFR(F3/F3) pulmonary arterial smooth muscle cells. On exposure to chronic hypoxia, vascular remodeling of pulmonary arteries was blunted in βPDGFR(F3/F3) mice compared with wild-type littermates. These alterations led to protection from hypoxia-induced PAH and right ventricular hypertrophy.
Conclusions: By means of a genetic approach, our data provide definite evidence that the activated βPDGFR is a key contributor to pulmonary vascular remodeling and PAH. Selective disruption of PDGF-dependent phophatidyl-inositol-3-kinase and PLCγ activity is sufficient to abolish these pathogenic responses in vivo, identifying these signaling events as valuable targets for antiremodeling strategies in PAH.
Keywords: platelet-derived growth factor; pulmonary hypertension; vascular endothelial growth factor; vascular remodeling.
© 2015 American Heart Association, Inc.
Figures
Similar articles
-
Disruption of platelet-derived growth factor-dependent phosphatidylinositol 3-kinase and phospholipase Cγ 1 activity abolishes vascular smooth muscle cell proliferation and migration and attenuates neointima formation in vivo.J Am Coll Cardiol. 2011 Jun 21;57(25):2527-38. doi: 10.1016/j.jacc.2011.02.037. J Am Coll Cardiol. 2011. PMID: 21679854 Free PMC article.
-
Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases.Am J Respir Crit Care Med. 2011 Apr 15;183(8):1092-102. doi: 10.1164/rccm.200911-1663OC. Epub 2010 Dec 22. Am J Respir Crit Care Med. 2011. PMID: 21177885
-
NMDA-Type Glutamate Receptor Activation Promotes Vascular Remodeling and Pulmonary Arterial Hypertension.Circulation. 2018 May 29;137(22):2371-2389. doi: 10.1161/CIRCULATIONAHA.117.029930. Epub 2018 Feb 14. Circulation. 2018. PMID: 29444988
-
Targeting of platelet-derived growth factor signaling in pulmonary arterial hypertension.Handb Exp Pharmacol. 2013;218:381-408. doi: 10.1007/978-3-642-38664-0_16. Handb Exp Pharmacol. 2013. PMID: 24092349 Review.
-
PDGF signaling in pulmonary arterial hypertension.J Clin Invest. 2005 Oct;115(10):2691-4. doi: 10.1172/JCI26593. J Clin Invest. 2005. PMID: 16200204 Free PMC article. Review.
Cited by
-
IMPAHCT: A randomized phase 2b/3 study of inhaled imatinib for pulmonary arterial hypertension.Pulm Circ. 2024 Mar 25;14(1):e12352. doi: 10.1002/pul2.12352. eCollection 2024 Jan. Pulm Circ. 2024. PMID: 38532768 Free PMC article.
-
The Platelet-Derived Growth Factor Pathway in Pulmonary Arterial Hypertension: Still an Interesting Target?Life (Basel). 2022 Apr 29;12(5):658. doi: 10.3390/life12050658. Life (Basel). 2022. PMID: 35629326 Free PMC article. Review.
-
mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target.Int J Mol Sci. 2021 Feb 21;22(4):2144. doi: 10.3390/ijms22042144. Int J Mol Sci. 2021. PMID: 33670032 Free PMC article. Review.
-
Macrophage-derived PDGF-B induces muscularization in murine and human pulmonary hypertension.JCI Insight. 2021 Mar 22;6(6):e139067. doi: 10.1172/jci.insight.139067. JCI Insight. 2021. PMID: 33591958 Free PMC article.
-
Endothelial Twist1-PDGFB signaling mediates hypoxia-induced proliferation and migration of αSMA-positive cells.Sci Rep. 2020 May 5;10(1):7563. doi: 10.1038/s41598-020-64298-5. Sci Rep. 2020. PMID: 32371931 Free PMC article.
References
-
- Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. - DOI - PubMed
-
- Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(suppl 1):S20–S31. doi: 10.1016/j.jacc.2009.04.018. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
