

Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection
- PMID: 25745066
- PMCID: PMC4856222
- DOI: 10.1126/science.1258867
Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection
Abstract
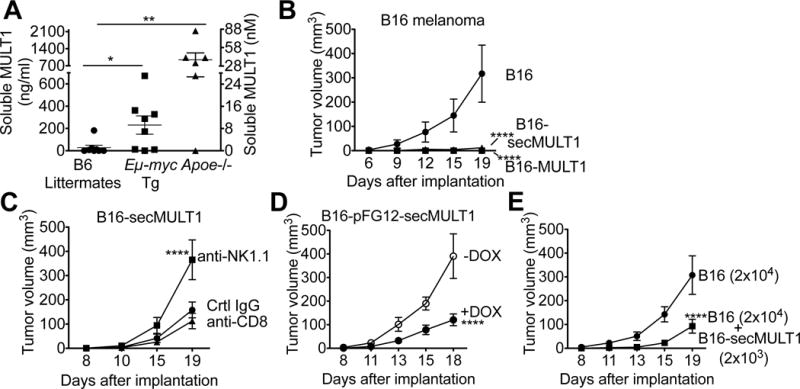
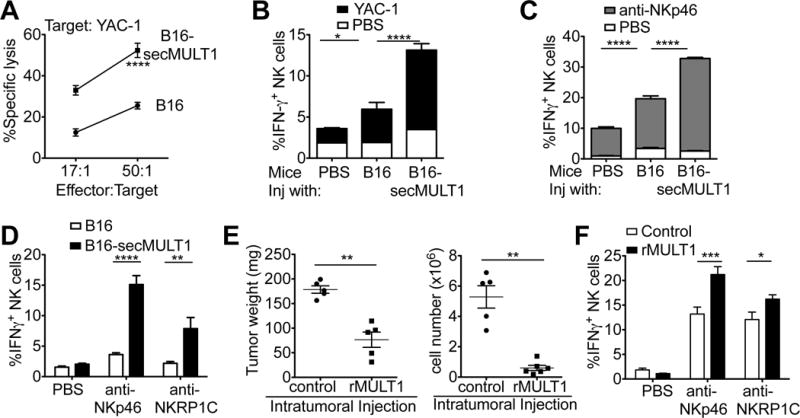
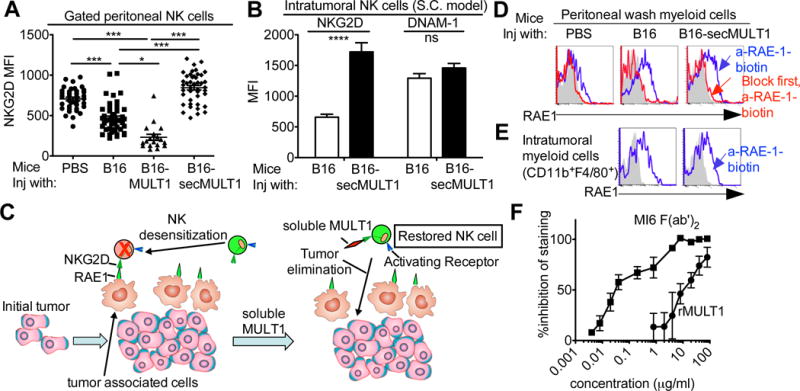
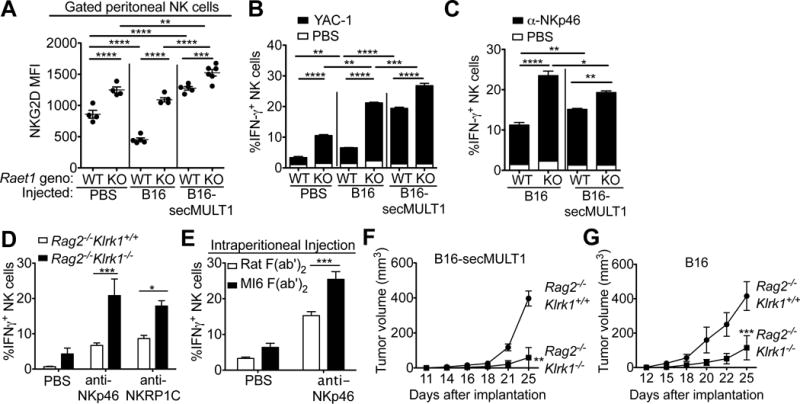
Immune cells, including natural killer (NK) cells, recognize transformed cells and eliminate them in a process termed immunosurveillance. It is thought that tumor cells evade immunosurveillance by shedding membrane ligands that bind to the NKG2D-activating receptor on NK cells and/or T cells, and desensitize these cells. In contrast, we show that in mice, a shed form of MULT1, a high-affinity NKG2D ligand, causes NK cell activation and tumor rejection. Recombinant soluble MULT1 stimulated tumor rejection in mice. Soluble MULT1 functions, at least in part, by competitively reversing a global desensitization of NK cells imposed by engagement of membrane NKG2D ligands on tumor-associated cells, such as myeloid cells. The results overturn conventional wisdom that soluble ligands are always inhibitory and suggest a new approach for cancer immunotherapy.
Copyright © 2015, American Association for the Advancement of Science.
Figures
Comment in
-
Immunotherapy: Soluble ligands--a new approach to cancer therapy.Nat Rev Clin Oncol. 2015 Jun;12(6):311. doi: 10.1038/nrclinonc.2015.60. Epub 2015 Mar 24. Nat Rev Clin Oncol. 2015. PMID: 25801813 No abstract available.
-
Immunology. MULT1plying cancer immunity.Science. 2015 Apr 3;348(6230):45-6. doi: 10.1126/science.aaa9842. Science. 2015. PMID: 25838370 No abstract available.
-
Shed NKG2D ligand boosts NK cell immunity.Cell Res. 2015 Jun;25(6):651-2. doi: 10.1038/cr.2015.41. Epub 2015 Apr 7. Cell Res. 2015. PMID: 25849247 Free PMC article.
-
Distinct mechanisms of tumor resistance to NK killing: of mice and men.Immunity. 2015 Apr 21;42(4):605-6. doi: 10.1016/j.immuni.2015.04.007. Immunity. 2015. PMID: 25902479 Free PMC article.
Similar articles
-
Shed NKG2D ligand boosts NK cell immunity.Cell Res. 2015 Jun;25(6):651-2. doi: 10.1038/cr.2015.41. Epub 2015 Apr 7. Cell Res. 2015. PMID: 25849247 Free PMC article.
-
Distinct mechanisms of tumor resistance to NK killing: of mice and men.Immunity. 2015 Apr 21;42(4):605-6. doi: 10.1016/j.immuni.2015.04.007. Immunity. 2015. PMID: 25902479 Free PMC article.
-
Critical role of the NKG2D receptor for NK cell-mediated control and immune escape of B-cell lymphoma.Eur J Immunol. 2015 Sep;45(9):2593-601. doi: 10.1002/eji.201445375. Epub 2015 Jul 7. Eur J Immunol. 2015. PMID: 26151313
-
Generation of soluble NKG2D ligands: proteolytic cleavage, exosome secretion and functional implications.Scand J Immunol. 2013 Aug;78(2):120-9. doi: 10.1111/sji.12072. Scand J Immunol. 2013. PMID: 23679194 Review.
-
Murine NKG2D ligands: "double, double toil and trouble".Mol Immunol. 2009 Mar;46(6):1011-9. doi: 10.1016/j.molimm.2008.09.035. Epub 2008 Dec 10. Mol Immunol. 2009. PMID: 19081632 Free PMC article. Review.
Cited by
-
Targeting NK Cells for Anticancer Immunotherapy: Clinical and Preclinical Approaches.Front Immunol. 2016 Apr 21;7:152. doi: 10.3389/fimmu.2016.00152. eCollection 2016. Front Immunol. 2016. PMID: 27148271 Free PMC article. Review.
-
NKG2D Receptor and Its Ligands in Host Defense.Cancer Immunol Res. 2015 Jun;3(6):575-82. doi: 10.1158/2326-6066.CIR-15-0098. Cancer Immunol Res. 2015. PMID: 26041808 Free PMC article. Review.
-
The cancer-natural killer cell immunity cycle.Nat Rev Cancer. 2020 Aug;20(8):437-454. doi: 10.1038/s41568-020-0272-z. Epub 2020 Jun 24. Nat Rev Cancer. 2020. PMID: 32581320 Review.
-
Sepsis inhibits tumor growth in mice with cancer through Toll-like receptor 4-associated enhanced Natural Killer cell activity.Oncoimmunology. 2019 Jul 19;8(11):e1641391. doi: 10.1080/2162402X.2019.1641391. eCollection 2019. Oncoimmunology. 2019. PMID: 31646090 Free PMC article.
-
Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade.J Clin Invest. 2018 Oct 1;128(10):4654-4668. doi: 10.1172/JCI99317. Epub 2018 Sep 10. J Clin Invest. 2018. PMID: 30198904 Free PMC article.
References
-
- Raulet DH. Nat Rev Immunol. 2003;3:781–790. - PubMed
-
- Chitadze G, Bhat J, Lettau M, Janssen O, Kabelitz D. Scandinavian journal of immunology. 2013;78:120–129. - PubMed
-
- Groh V, Wu J, Yee C, Spies T. Nature. 2002;419:734–738. - PubMed
-
- Song H, Kim J, Cosman D, Choi I. Cell Immunol. 2006;239:22–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
