

Differential expression of HERV-K (HML-2) proviruses in cells and virions of the teratocarcinoma cell line Tera-1
- PMID: 25746218
- PMCID: PMC4379556
- DOI: 10.3390/v7030939
Differential expression of HERV-K (HML-2) proviruses in cells and virions of the teratocarcinoma cell line Tera-1
Abstract
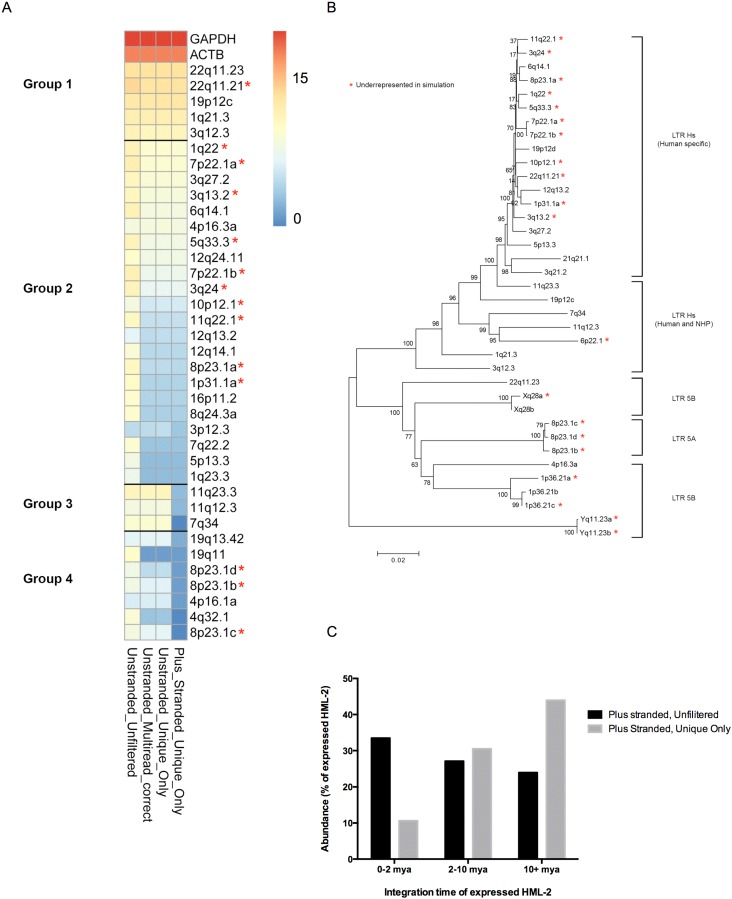
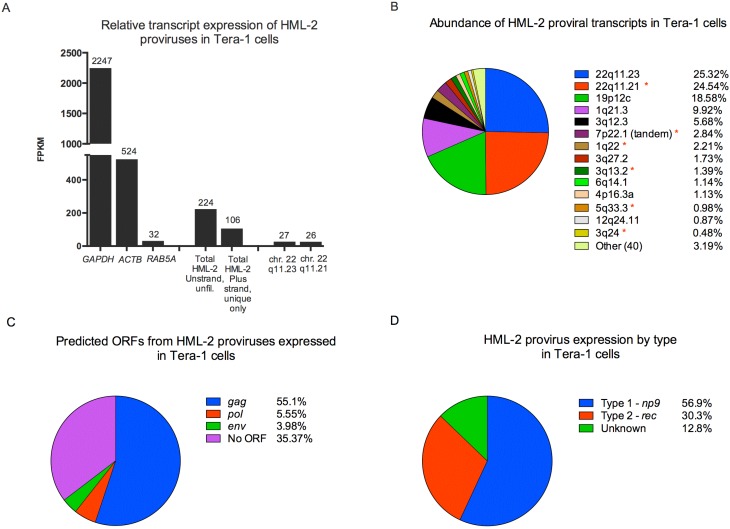
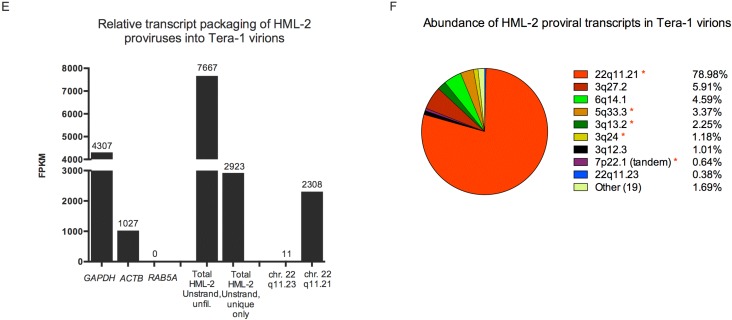
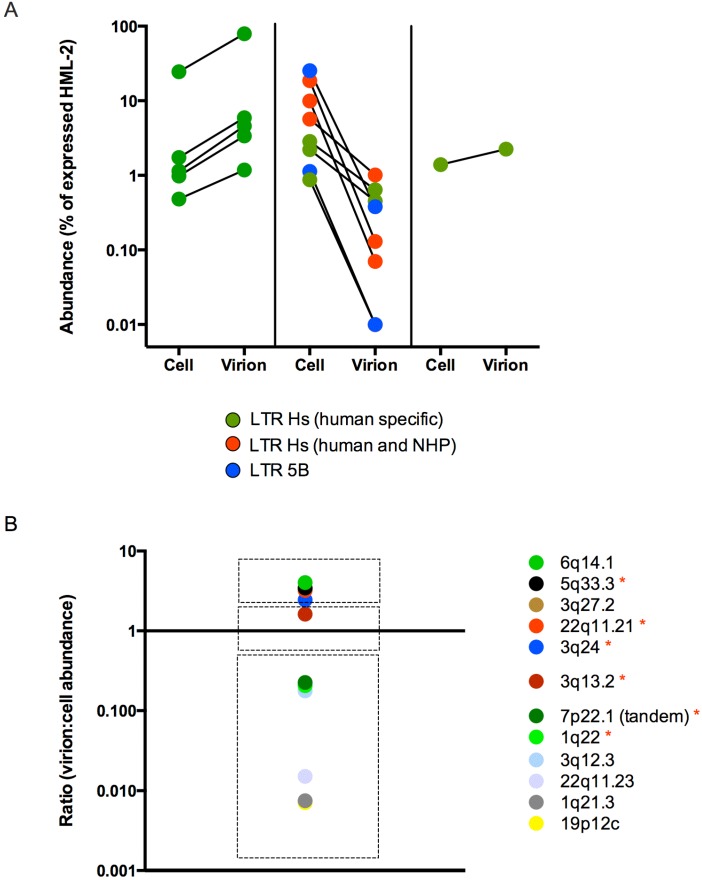
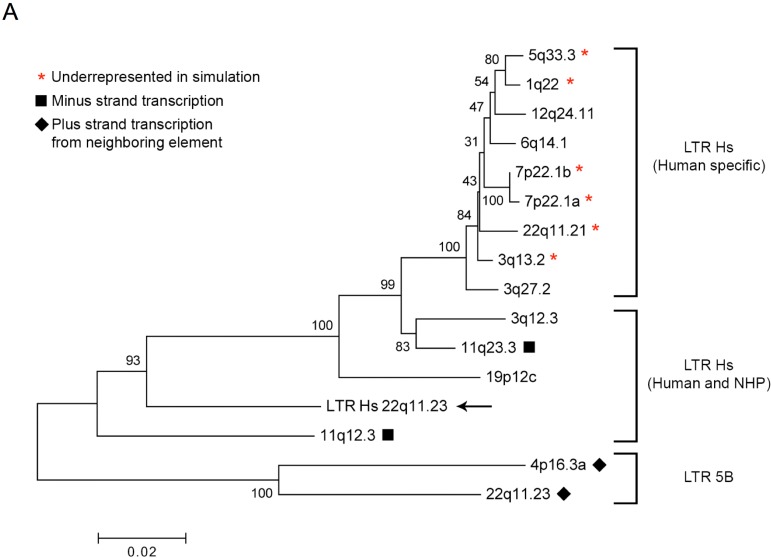
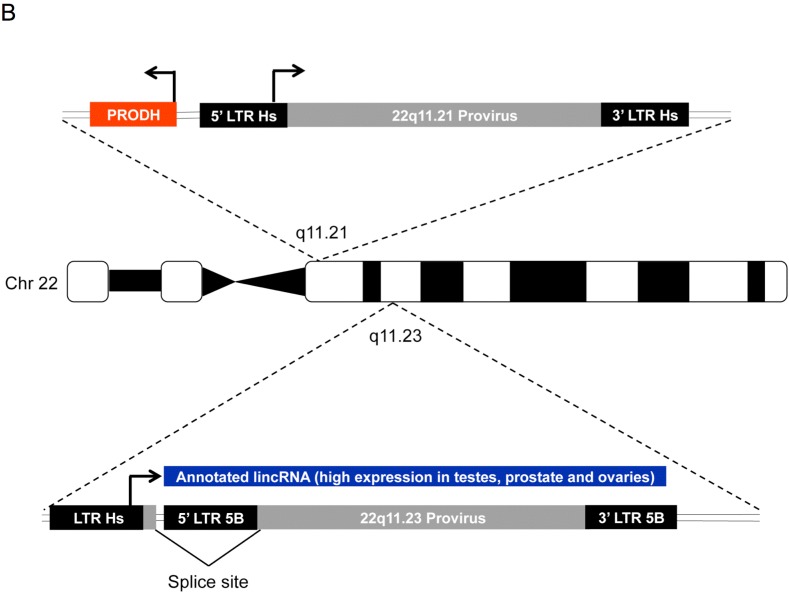
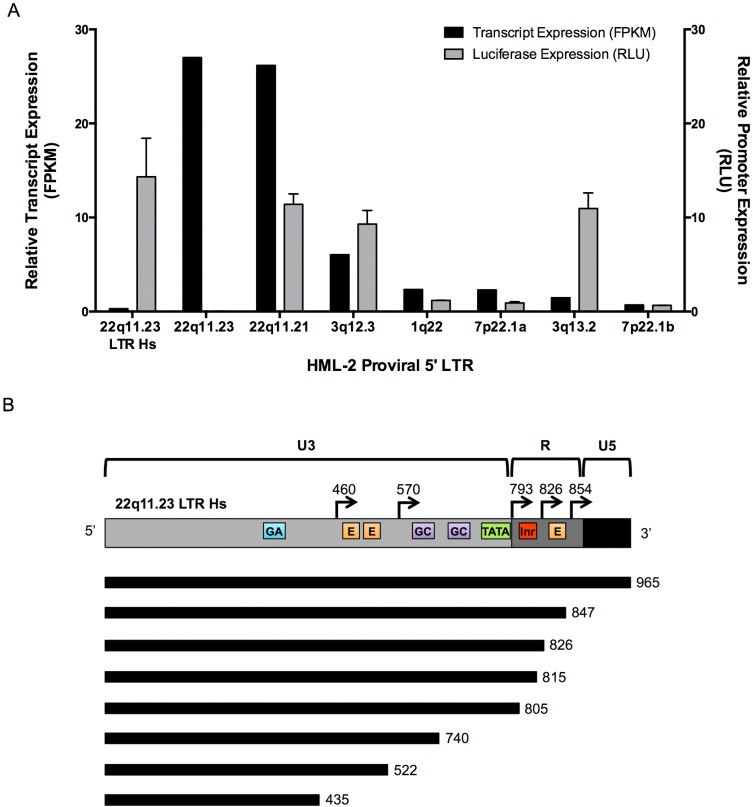
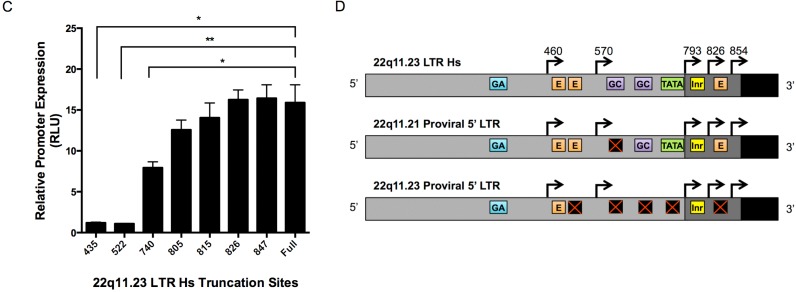
Human endogenous retrovirus (HERV-K (HML-2)) proviruses are among the few endogenous retroviral elements in the human genome that retain coding sequence. HML-2 expression has been widely associated with human disease states, including different types of cancers as well as with HIV-1 infection. Understanding of the potential impact of this expression requires that it be annotated at the proviral level. Here, we utilized the high throughput capabilities of next-generation sequencing to profile HML-2 expression at the level of individual proviruses and secreted virions in the teratocarcinoma cell line Tera-1. We identified well-defined expression patterns, with transcripts emanating primarily from two proviruses located on chromosome 22, only one of which was efficiently packaged. Interestingly, there was a preference for transcripts of recently integrated proviruses, over those from other highly expressed but older elements, to be packaged into virions. We also assessed the promoter competence of the 5' long terminal repeats (LTRs) of expressed proviruses via a luciferase assay following transfection of Tera-1 cells. Consistent with the RNASeq results, we found that the activity of most LTRs corresponded to their transcript levels.
Figures
References
-
- Boeke J.D., Stoye J.S. Retrotransposons, Endogenous Retroviruses, and the Evolution of Retroelements. In: Coffin J.M., Hughes S.H., Varmus H.E., editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY, USA: 1997. pp. 343–435. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
