

ChIP-nexus enables improved detection of in vivo transcription factor binding footprints
- PMID: 25751057
- PMCID: PMC4390430
- DOI: 10.1038/nbt.3121
ChIP-nexus enables improved detection of in vivo transcription factor binding footprints
Abstract
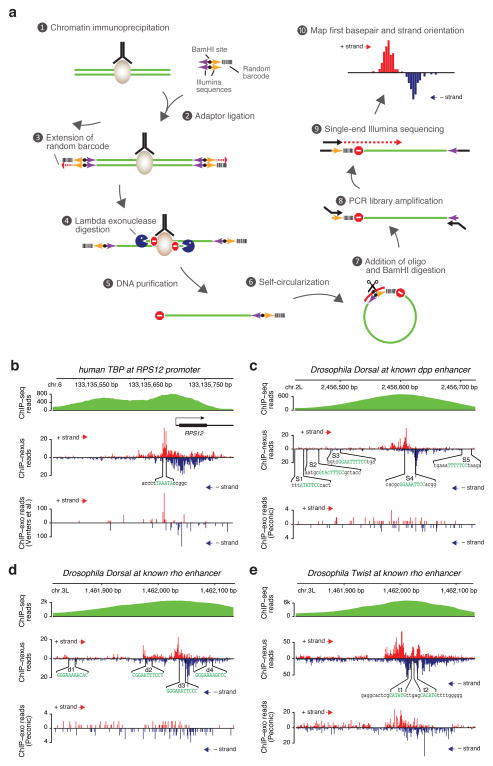
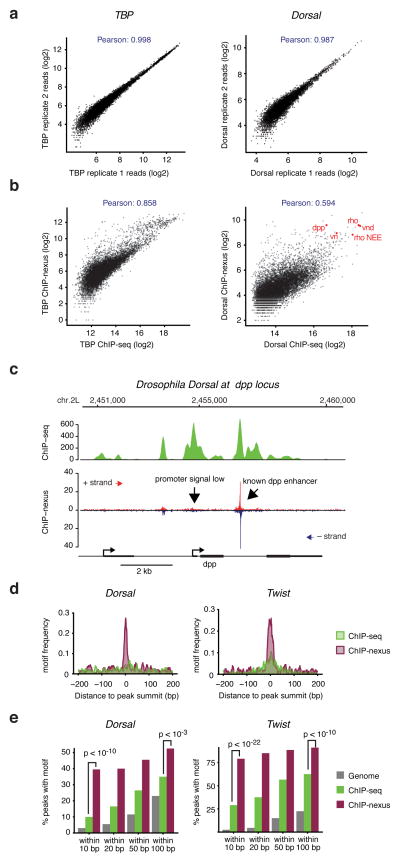
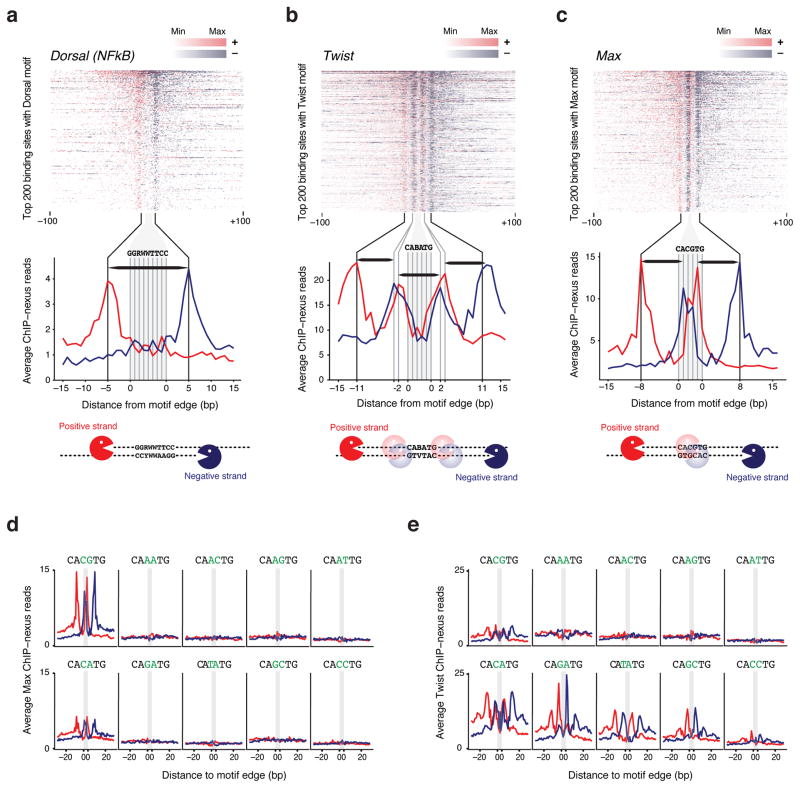
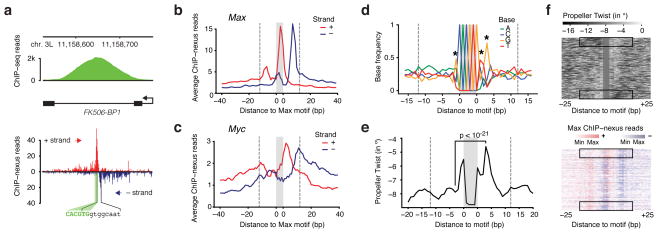
Understanding how eukaryotic enhancers are bound and regulated by specific combinations of transcription factors is still a major challenge. To better map transcription factor binding genome-wide at nucleotide resolution in vivo, we have developed a robust ChIP-exo protocol called ChIP-nexus (chromatin immunoprecipitation experiments with nucleotide resolution through exonuclease, unique barcode and single ligation), which utilizes an efficient DNA self-circularization step during library preparation. Application of ChIP-nexus to four proteins--human TBP and Drosophila NFkB, Twist and Max--shows that it outperforms existing ChIP protocols in resolution and specificity, pinpoints relevant binding sites within enhancers containing multiple binding motifs, and allows for the analysis of in vivo binding specificities. Notably, we show that Max frequently interacts with DNA sequences next to its motif, and that this binding pattern correlates with local DNA-sequence features such as DNA shape. ChIP-nexus will be broadly applicable to the study of in vivo transcription factor binding specificity and its relationship to cis-regulatory changes in humans and model organisms.
Figures
References
-
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
