

De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy
- PMID: 25751627
- PMCID: PMC4380508
- DOI: 10.1038/ng.3239
De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy
Abstract
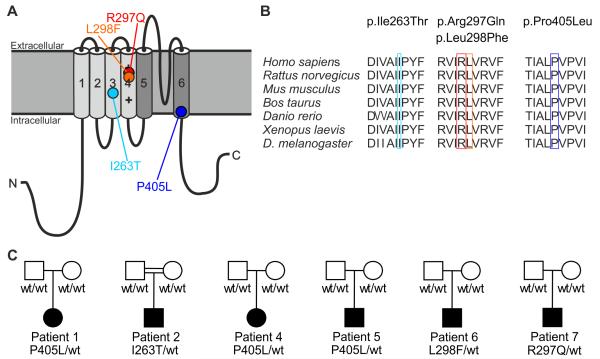
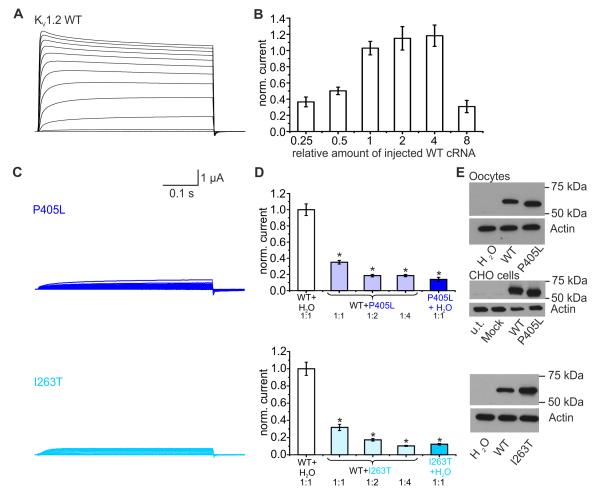
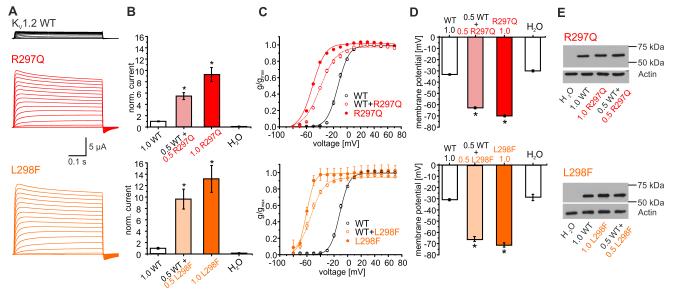
Epileptic encephalopathies are a phenotypically and genetically heterogeneous group of severe epilepsies accompanied by intellectual disability and other neurodevelopmental features. Using next-generation sequencing, we identified four different de novo mutations in KCNA2, encoding the potassium channel KV1.2, in six isolated patients with epileptic encephalopathy (one mutation recurred three times independently). Four individuals presented with febrile and multiple afebrile, often focal seizure types, multifocal epileptiform discharges strongly activated by sleep, mild to moderate intellectual disability, delayed speech development and sometimes ataxia. Functional studies of the two mutations associated with this phenotype showed almost complete loss of function with a dominant-negative effect. Two further individuals presented with a different and more severe epileptic encephalopathy phenotype. They carried mutations inducing a drastic gain-of-function effect leading to permanently open channels. These results establish KCNA2 as a new gene involved in human neurodevelopmental disorders through two different mechanisms, predicting either hyperexcitability or electrical silencing of KV1.2-expressing neurons.
Figures
Comment in
-
Maintaining the balance: both gain- and loss-of-function KCNA2 mutants cause epileptic encephalopathy.Clin Genet. 2015 Aug;88(2):137-9. doi: 10.1111/cge.12615. Epub 2015 Jun 3. Clin Genet. 2015. PMID: 25997620 No abstract available.
References
-
- Capovilla G, Wolf P, Beccaria F, Avanzini G. The history of the concept of epileptic encephalopathy. Epilepsia. 2013;54(Suppl 8):2–5. - PubMed
-
- Guerrini R, Pellock JM. Age-related epileptic encephalopathies. Handb Clin Neurol. 2012;107:179–93. - PubMed
-
- Nava C, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet. 2014;46:640–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
