

Development of a high-resolution NGS-based HLA-typing and analysis pipeline
- PMID: 25753671
- PMCID: PMC4477639
- DOI: 10.1093/nar/gkv184
Development of a high-resolution NGS-based HLA-typing and analysis pipeline
Abstract
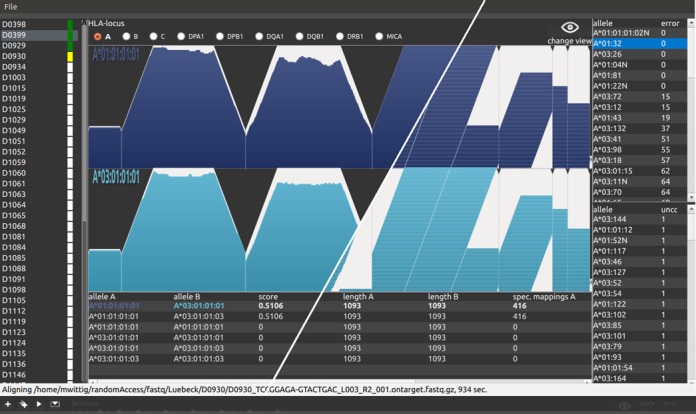
The human leukocyte antigen (HLA) complex contains the most polymorphic genes in the human genome. The classical HLA class I and II genes define the specificity of adaptive immune responses. Genetic variation at the HLA genes is associated with susceptibility to autoimmune and infectious diseases and plays a major role in transplantation medicine and immunology. Currently, the HLA genes are characterized using Sanger- or next-generation sequencing (NGS) of a limited amplicon repertoire or labeled oligonucleotides for allele-specific sequences. High-quality NGS-based methods are in proprietary use and not publicly available. Here, we introduce the first highly automated open-kit/open-source HLA-typing method for NGS. The method employs in-solution targeted capturing of the classical class I (HLA-A, HLA-B, HLA-C) and class II HLA genes (HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, HLA-DPB1). The calling algorithm allows for highly confident allele-calling to three-field resolution (cDNA nucleotide variants). The method was validated on 357 commercially available DNA samples with known HLA alleles obtained by classical typing. Our results showed on average an accurate allele call rate of 0.99 in a fully automated manner, identifying also errors in the reference data. Finally, our method provides the flexibility to add further enrichment target regions.
© The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

References
-
- Horton R., Wilming L., Rand V., Lovering R.C., Bruford E.A., Khodiyar V.K., Lush M.J., Povey S., Talbot C.C., Jr, Wright M.W., et al. Gene map of the extended human MHC. Nat. Rev. Genet. 2004;5:889–899. - PubMed
-
- Thorsby E., Lie B.A. HLA associated genetic predisposition to autoimmune diseases: genes involved and possible mechanisms. Transpl. Immunol. 2005;14:175–182. - PubMed
-
- Flomenberg N., Baxter-Lowe L.A., Confer D., Fernandez-Vina M., Filipovich A., Horowitz M., Hurley C., Kollman C., Anasetti C., Noreen H., et al. Impact of HLA class I and class II high-resolution matching on outcomes of unrelated donor bone marrow transplantation: HLA-C mismatching is associated with a strong adverse effect on transplantation outcome. Blood. 2004;104:1923–1930. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
